
ABSTRACT
Sepsis-induced immune dysfunctions are likely to impact on malignant tumor growth. Sequential sepsis-then-cancer models of tumor transplantation in mice recovering from sepsis have shown that the post-septic immunosuppressive environment was able to promote tumor growth. We herein addressed the impact of sepsis on pre-established malignancy in a reverse cancer-then sepsis experimental model. Mice previously inoculated with MCA205 fibrosarcoma cells were subjected to septic challenges by polymicrobial peritonitis induced by cecal ligation and puncture or endotoxinic shock. The anti-tumoral immune response was assessed through the distribution of tumor-infiltrating immune cells, as well as the functions of cytotoxic cells. As compared to sham surgery, polymicrobial sepsis dampened malignant tumor growth in wild-type (WT) mice, but neither in Toll-like receptor 4 (Tlr4)−/- nor in Myd88−/- mice. Similar tumor growth inhibition was observed following a LPS challenge in WT mice, suggesting a regulatory role of Tlr4 in this setting. The low expression of MHC class 1 onto MCA205 cells suggested the involvement of Natural Killer (NK) cells in sepsis-induced tumor inhibition. Septic insults applied to mice with cancer promoted the main anti-tumoral NK functions of IFNγ production and degranulation. The anti-tumoral properties of NK cells obtained from septic mice were exacerbated when cultured with MHC1low MCA205 or YAC-1 cells. These results suggest that sepsis may harbor dual effects on tumor growth depending on the sequential experimental model. When applied in mice with cancer, sepsis prevents tumor growth in a Tlr4-dependent manner by enhancing the anti-tumoral functions of NK cells.
Introduction
Sepsis remains a frequent and dreaded complication in vulnerable patients with malignancies. It is noteworthy that the short-term survival to sepsis has dramatically improved in cancer patients, raising new questions about their long-term outcome and the particular impact of severe bacterial infections on the oncological prognosis.Citation1 Sepsis is a complex clinical syndrome currently viewed as an early dysregulated inflammatory response followed by a complex immunosuppressive response combining innate and adaptive immune defects.Citation2 Once limited to anti-infective immune responses, the extension of sepsis-induced immune dysfunctions to the pathophysiology of cardiovascular diseases and malignancies has become a major field of interest.
The immune system plays a major role in the control of tumor development through the recognition and destruction of transforming cells and dying tumor cells. Chronic inflammation and some chronic infectious diseases are well known to promote oncogenesis and tumor progression mediated in part by modulation of tumor immune surveillance, but the role of acute inflammation and severe bacterial infections on cancer remains largely unclear.Citation3 Tumor immune surveillance is under the control of several innate and adaptive immune cell types that are likely to be affected by sepsis, and it is noteworthy that immune defects induced by sepsis and cancer share remarkable similarities, both at early and late stages of diseases.Citation4,Citation5 Following a septic insult, the acute inflammatory response and the subsequent impairment of immune responses can modify the tumoral immune microenvironment and may thereby promote or conversely inhibit tumor growth.Citation6–Citation10
In order to address the interactions between cancer and sepsis, we previously established a double-hit experimental model in which mice that survived a primary polymicrobial sepsis induced by cecal ligation and puncture (CLP) were subjected to subcutaneous fibrosarcoma cell inoculation.Citation11 As compared to sham-operated controls, post-septic mice exhibited accelerated tumor growth that could be linked to a Tlr4-dependent expansion of granulocytic myeloid-derived suppressor cells (MDSC). However, the sepsis-then-cancer sequence does not recapitulate the most common clinical situation of severe infectious complications occurring in cancer patients. In order to address the impact of sepsis on pre-established malignancy, we investigated a reverse sequential model in which mice were first subjected to malignant fibrosarcoma cell inoculation. Mice with cancer were secondarily subjected to polymicrobial sepsis or endotoxinic shock. In this cancer-then-sepsis model, sepsis rather inhibited tumor growth in a Tlr4-dependent manner through enhanced anti-tumoral functions of NK cells.
Materials and methods
Mice
Eight- to 12-week old mice of C57BL/6J background were used in all experiments. Wild-type (WT) mice were purchased from Janvier Laboratories. Tlr2−/-, Tlr4−/- and MyD88−/- mice were kind gifts from Prof. Shizuo Akira (Osaka University, Japan). All animals were maintained in the specific pathogen-free animal facility of the Cochin Institute. Experiments were conducted in compliance with European animal welfare regulations. The study was approved by the Paris Descartes University ethics committee for animal experimentation.
Tumor inoculation
We used the 3-methylcholanthrene-induced fibrosarcoma cell line MCA205,Citation12 which was kindly provided by Prof. Antoine Tesnière (INSERM U1138, Centre de Recherche des Cordeliers, Paris, France). Cells were cultured under semi-confluent conditions in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 5% fetal calf serum (FCS, Deutscher), 1% penicillin/streptomycin, 1% L-Glutamin and 1% HEPES (Gibco) at 37°C under 5% CO2. Cells were harvested for experiments by a brief application of trypsin (Gibco) and washed once with Phosphate-Buffered Saline (PBS). We checked that this cell line was never reported misidentified in both ICLAC and NCBI BioSample databases.Citation13,Citation14
We used two different models of subcutaneous or intravenous MCA205 tumor inoculation. Mice were subjected to subcutaneous (s.c.) injection of 5 × 104 MCA205 cells into the right flank, resulting in localized tumor growth without any metastatic dissemination. Tumors became detectable mostly after 7 days from inoculation. Tumors were measured using a caliper and the volume was determined using the following ellipsoid volume approximation formula: (length×width2) × 0.52. Tumor growth was expressed by tumor volumes and absolute variations of tumor volume between Dx + 7 and Dx. Alternatively mice were subjected to retro-orbital intravenous (i.v.) inoculation of 105 MCA205 cells under short-term general anesthesia.
Sepsis models
We used the classical model of polymicrobial sepsis induced by CLP.Citation15 Mice were anaesthetized with a combination of Ketamin/Xylazin (100 µg/10 µg per g bodyweight). The cecum was exposed, ligatured at its external third and punctured through with a 26-gauge needle. Incision was sutured in layers and animals were resuscitated with an intra-peritoneal injection of 1mL saline. Control animals were subjected to sham surgery by laparotomy only without CLP. Six hours following surgery and then every 12 hours during 3 days, mice received intra-peritoneal fluid resuscitation and injection of painkillers (0.5mL saline + buprenorphine 0.1µg per g bodyweight). We also used a surrogate model of sepsis through endotoxinic shock induced by intraperitoneal (i.p.) injection of lipopolysaccharide (LPS) at the dose of 5 µg per g bodyweight (O55:B5 Escherichia coli LPS, Sigma-Aldrich). In one experiment, mice were NK-depleted by an i.p. injection of 250 µg anti-NK1.1 antibody (InVivoMab PK136, Bio X Cell) administered 24h before the septic challenge and repeated once three days later.
Assessment of lung metastasis
Mice were subjected to intratracheal injection of a 15-percent solution of India ink. Lungs were removed and washed, and fixed overnight in Fekete’s solution. Tumor nodules show up white. For histological analysis, whole lungs were successively fixed in 4% paraformaldehyde (Euromedex), dehydrated in ethanol, embedded in paraffin and cut into 4-µm sections. Sections were stained with hematoxylin-eosin for tissue examination. For quantitative analysis, images were acquired with an AZ100 Nikon microscope and analysed with NIS-Elements Basic Research Software (Nikon). The tumoral infiltration of the right lung expressed as a proportion of the whole lung surface was assessed in 3 random sections from the same mouse using ImageJ software
RNA isolation and real-time polymerase chain reaction (rt-PCR)
Tumors were surgically removed under sterile conditions, frozen and ground on dry ice. Samples were lysed with TissueLyser II (Qiagen) using stainless steel beads. RNA was extracted and purified with the PureLink RNA Mini Kit (Invitrogen) and cDNA was synthesized using Superscript Reverse Transcriptase (Invitrogen). Two steps of DNase were carried out using PureLink DNase (Invitrogen) and DNase Amplification Grade (Invitrogen) following the manufacturer’s instructions. Levels of the mRNA from the target genes Prf1, Gzmb, Ifng, Tnf, Il10, Tgfb1, Il12a and Il12b as well as the housekeeping genes Sdha and Actin were quantified by real-time PCR using LightCycler® 480 SYBR Green I Master (Roche) and gene-specific primers (Eurogentec). Gene expression was adjusted, normalized to housekeeping genes and expressed as normalized relative quantity (NRQ).
Determination of cytokine levels
TNFA and IL10 were quantified in supernatants of tumor lysates using Luminex multiplex assays (Bio-Plex200; Bio-Rad) according to the manufacturer’s instructions. Data were analysed using Bio-Plex Manager (Bio-Rad).
Single-cell suspension procedures
Mice were euthanized, spleens and tumors were surgically removed under sterile conditions and digested with 1 mg/mL collagenase D and 0.1 mg/mL DNase I (Sigma-Aldrich) for respectively 10 min and 20 min at 37°C before being mechanically dissociated and smashed through a 40 μm nylon strainer in PBS supplemented with 5% FCS, 0.1% NaN3 and 0,5% EDTA (Sigma-Aldrich). Red blood cells were removed using BD Pharm LyseTM lysis buffer and cells were enumerated with Accuri C6 before staining.
Cell-surface staining and flow cytometry
The following anti-mouse antibodies were used for FACS staining: CD45-AF700 (30F11, BioLegend), CD3ε-PerCP-Vio700 (145-2C11, Miltenyi Biotec), NK1.1-BV421 (PK136, BioLegend), CD8a-APC-Vio770 (53–6.7, Miltenyi Biotec), CD4-BV785 (GK1.5, BioLegend), CD11c-PeCy7 (HL3, BD Biosciences), Ly6G-APC (1A8, BD Biosciences), Ly6C-BV421 (AL-21, BD Biosciences), Tγ/δ-PE (GL3, BD Biosciences), CD19-FITC (1D3, BD Biosciences), CD11b-BV650 (M1/70, BioLegend), CD11b-FITC (M1/70.15.11.5, Miltenyi Biotec), F4/80-PE (REA126, Miltenyi Biotec), CD27-PE-Vio770 (REA499, Miltenyi Biotec), NKG2D-FITC (C7, BioLegend), CD25-APC (PC61, BioLegend), CD107a-BV711 (1D4B, BD Biosciences), IFNγ-PE (XMG1.2, BioLegend), Perforin-APC (S16009A, BioLegend), Granzyme B-FITC (GB11, BioLegend), H-2-FITC (M1/42, BioLegend) and Tlr4 (SA15-21, BioLegend) and their control isotype rat IgG2aκ (BioLegend).
Cells suspensions were first incubated with Fc-blocker anti-CD16/32 antibody (Miltenyi Biotech). Cells were stained with fluorescent antibodies for surface markers, and secondarily permeabilized with Cytofix/Cytoperm (BD Biosciences) and stained for intracellular molecules. Events were collected on a 4-laser Fortessa cytometer (BD Biosciences) and analyzed using Flowjo software. Myeloid and lymphoid subsets among CD45-positive hematopoietic cells were identified as previously described.Citation11 The geometric mean fluorescence intensity was used to quantify the antibody expression. The gating strategy to identify cytotoxic cell subsets was set up in spleen tissue and then applied to tumor tissue (figure S1). The assessment of activation and functions of intra-tumoral NK cells is described in the figure S2.
Degranulation assay
YAC-1 cells were kindly provided by Isabelle Munoz (Cochin Institute). Splenic NK cells from septic or non-septic mice were purified using an immunomagnetic procedure (NK Cell Isolation Kit, Miltenyi Biotech), reaching a purity > 90% of viable cells. 5 × 104 NK cells were plated with 5 × 104 MCA205 or YAC-1 cells (effector to target ratio 1:1) and 1 µl of anti-CD107a-BV711 (1D4B, BD Biosciences) per well. Brefeldin A (GolgiPlugTM BD Biosciences) was added one hour later. Cells were incubated for 4h and 18h at 37°C under 5% CO2, and then spun down and stained for intracellular molecules.
Statistical analysis
Continuous variables were expressed as boxplots or scatterplots or mean ± SD and compared using the Student t-test. P values < .05 indicated statistically significant differences.
Results
Polymicrobial sepsis inhibits tumor growth in mice with cancer
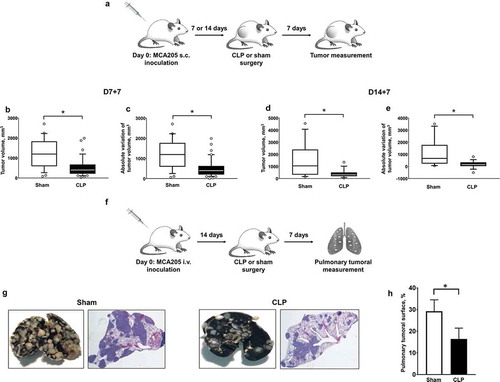
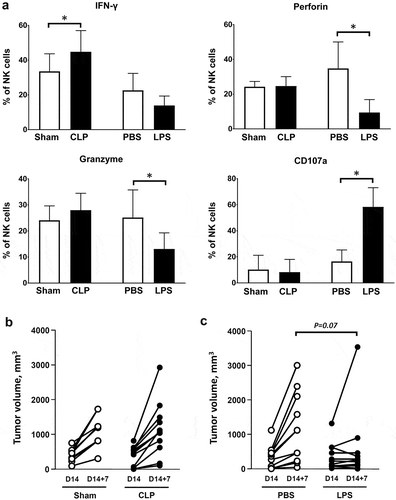
In order to address the impact of sepsis in hosts harboring cancer, we established double-hit experimental models (, )). WT mice were first subcutaneously inoculated with MCA205 cells, resulting in localized tumor growth. Tumors generally became detectable after 7 days from inoculation. Seven or 14 days after tumor inoculation, mice with cancer were subjected to polymicrobial peritonitis induced by CLP or to sham non-septic surgery. As compared to sham surgery, polymicrobial sepsis inhibited local tumor growth (–)). Alternatively, mice were first subjected to i.v. inoculation of MCA205 cells, thereby mimicking pulmonary metastatic disease. Polymicrobial sepsis also dramatically decreased the number and size of secondary pulmonary lesions (, )). Of note, mice with cancer did not display increased mortality to polymicrobial sepsis when compared to non-cancer counterparts (Figure S3).
Figure 1. Polymicrobial sepsis inhibits malignant tumor growth in mice with cancer. (a) Sequential local model with s.c. inoculation of MCA205 cells (day 0) followed by CLP-induced polymicrobial sepsis or sham surgery at days 7 or 14. Tumor volumes (b and d) and absolute variations of tumor volumes (c and e) were measured 7 days after surgery (D7 + 7 or D14 + 7, respectively). n = 15–20 mice per group. (f) Sequential metastatic model with i.v. inoculation of MCA205 tumor cells (day 0) followed 14 days later by CLP-induced polymicrobial sepsis or sham surgery. The pulmonary tumoral spread was measured 7 days after surgery. (g) Representative India ink-colored lungs and hematoxylin-eosin-stained lung sections in sham- and CLP-operated mice previously subjected to i.v. inoculation of MCA205 cells. The tumoral surface of the right lung was assessed in 3 random sections from the same mouse (h) (n = 3 mice per group). *p < .05.
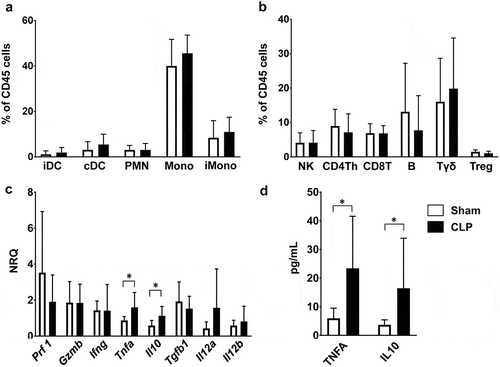
In order to understand the mechanisms that underlie the antitumoral effect of sepsis in this setting, we characterized the immune response within tumors in CLP- and sham-operated WT mice. At 24 hours after surgery, the distribution of intratumoral myeloid and lymphoid immune cells appeared quite similar between septic and non-septic mice (, )). In addition we assessed the whole expression of six representative cytokines within tumor tissues from septic and non-septic mice. CLP induced changes within the tumor inflammatory environment with increased expressions of Tnfα and il10 genes, consistent with increased concentrations of the TNFA and IL10 proteins in tumor lysate supernatants (, )).
Figure 2. Characteristics of sepsis-induced anti-tumoral immune response. Mice were subjected to CLP or sham surgery 14 days after s.c. MCA205 inoculation. Tumors were removed at 24 hours following surgery. (a and b) Distribution of myeloid and lymphoid subsets among viable CD45+ hematopoietic cells within tumors from sham-operated (white, n = 11) and CLP-operated (black, n = 10) mice. T helper lymphocytes (Th) are CD4+CD25−. T regulatory cells (Treg) are CD4+CD25+. (c) Expressions of Prf1, Gzmb, Ifng, Tnf, Il10, Tgfb1, Il12a and Il12b were quantified by RT-qPCR within tumors from sham-operated (white, n = 8) and CLP-operated (black, n = 12) mice. (d) TNFA and IL10 protein levels within supernatants of tumor homogenates obtained from sham-operated (white, n = 7) and post-CLP (black, n = 7) mice. NRQ, normalized relative quantity; iDC, inflammatory dendritic cells; cDC, conventional dendritic cells; PMN, polymorphonuclear cells; Mono, monocytes/macrophages; iMono, inflammatory monocytes/macrophages; Th, T helper; Treg, regulatory T cells. *p < .05.
Sepsis-induced tumor inhibition relies on tlr4-dependent signaling
The CLP model is associated with bacteremic dissemination of both Gram-negative and Gram-positive bacteria (data not shown). Following recognition of pathogen-associated molecular patterns, TLRs are major regulators of the immune response of sepsis. In order to address the role of TLR-dependent signaling in sepsis-induced tumor inhibition, we replicated the double-hit model of local tumor development followed by polymicrobial sepsis in mice deficient for Tlr2, Tlr4 or their mutual adaptive protein MyD88. Polymicrobial sepsis applied to Tlr2−/- mice with cancer resulted in tumor growth inhibition similar to that observed in WT littermates. In contrast sepsis-induced tumor inhibition was prevented in Tlr4−/- and MyD88−/- mice (, )). Likewise, polymicrobial sepsis-induced inhibition of lung metastasis was abrogated in Tlr4−/- mice (figure S4).
Figure 3. Sepsis-induced tumor growth inhibition is dependent on Tlr4 signaling. The sequential model of s.c. MCA205 inoculation followed by sham (white) or CLP (black) surgery at day 14 was applied to Tlr2−/- mice (n = 11 and 8 respectively), Tlr4−/- mice (n = 23 and 17 respectively), and MyD88−/- mice (n = 10 and 14 respectively). Tumor volumes (a) and absolute tumor variations (b) were measured 7 days after surgery. WT mice were subjected to s.c. MCA205 inoculation followed by PBS (white, n = 32), heat-killed Staphylococcus aureus (HKSA) (grey, n = 12) or LPS (black, n = 30) i.p. injection at day 14. Tumor volumes (c) and absolute tumor variations (d) were measured 7 days after i.p. injection. *p < .05.
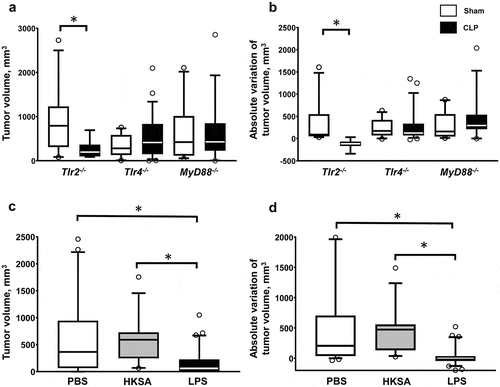
To confirm the implication of TLR signaling in this phenomenon, WT mice with cancer were challenged with Tlr2 and Tlr4 agonists as surrogate models of sepsis. Whereas i.p. administration of heat-killed Staphylococcus aureus (HKSA) at the dose of 108 or 109 CFU did not impact on tumor development, endotoxinic shock induced by i.p. injection of LPS resulted in inhibition, and occasionally regression, in tumor growth (, )). The LPS challenge did not modify T and NK cell counts but resulted in a dramatic increase in Il12a and Il12b gene expressions within tumor tissues (Figure S5). Of note, the growth of MCA205 cells in vitro was not altered when cultured in the presence of HKSA or LPS (Figure S6). Altogether, these data point to a regulatory role of Tlr4 signaling in shaping antitumoral immune responses during sepsis.
Sepsis-induced modulation of tumor-associated NK functions
Cytotoxic NK and T8 lymphocytes are the main tumoricidal effector cells through the release of cytotoxic molecules such as perforin and granzyme B into target cells, as well as production of IFNγ to sustain the antitumoral immune responses. The dramatic antitumoral effect observed in septic animals suggests that acute bacterial challenges may promote the antitumoral functions of cytotoxic cells. In line with this hypothesis, we observed that MCA205 cells exhibited low MHC class I expression similar to that of the classical NK-sensitive YAC-1 cell line, suggesting that they could also be targeted by NK cells ()).
Figure 4. Tumor-associated NK subsets. (a) Major histocompatibility complex class 1 expression on MCA205 and YAC-1 cell lines and on splenocytes from naive mice (positive control) stained with anti-H2 antibody. (b–d) Mice were subjected to s.c. inoculation of MCA205 tumor cells followed followed 14 days later by a septic insult (sham surgery or CLP, or PBS or LPS i.p. challenge). Tumors were removed at 24 hours following the septic challenge to assess the distribution of intratumoral NK subsets based on CD11b and CD27 expressions (b) and the membrane expressions of NKG2D (c) and CD25 (d) on intratumoral NK cells. 6–11 mice/group from two independent experiments. MFI, median fluorescence intensity. *p < .05.
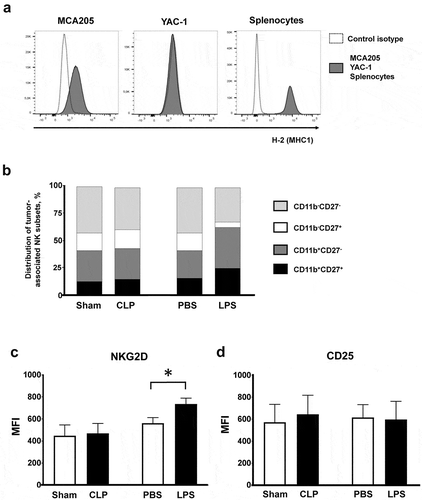
NK cells can be distributed into various functional subsets based on CD11b and CD27 membrane expressions. LPS administration resulted in mild expansion of the tumor-associated CD11b+CD27+ and CD11b+CD27− cytotoxic subsets ()) along with increased expression of the NKG2D activating receptor ()) without any subset specificity (data not show). There was no difference in the expression of CD25 ()). Tumor-associated NK cells retained very low Tlr4 membrane expression in all control and septic conditions (Figure S7). NK cells appeared evenly distributed within tumor tissues (data not shown).
Tumor-associated NK cells obtained from CLP-operated mice exhibited increased intracellular expression of IFNγ. NK cells from LPS-challenged mice displayed intracellular depletion in both perforin and granzyme B while the outer membrane expression of the degranulation marker CD107a (lysosomal-associated membrane protein-1) dramatically increased, suggesting that degranulation had already occurred ()). Regardless of the type of insult (CLP or LPS), a septic challenge did not induce significant changes in the functions of CD8+ T lymphocytes (Figure S8). In order to address the functional role of NK cells in sepsis-induced inhibition in tumor growth, mice with cancer were NK-depleted at the time of CLP/sham surgery or LPS/PBS challenge. A single i.p. injection of 250µg anti-NK1.1 antibody resulted in 85-percent depletion of tumor-associated NK cells within 24h (data not shown). Of note, NK depletion did not alter the survival of CLP-operated mice (day-7 mortality rate 45%). NK depletion restored full tumor growth in CLP-operated mice ()), but had little or no impact on LPS-induced tumor inhibition ()).
Figure 5. Functional role of tumor-associated NK cells in sepsis-induced tumor inhibition. Mice were subjected to s.c. inoculation of MCA205 tumor cells followed followed 14 days later by a septic insult (sham surgery or CLP, or PBS or LPS i.p. challenge). (a) Tumors were removed at 24 hours following the septic challenge for the assessment of intracellular expression of IFNγ, perforin, granzyme B and outer membrane expression of CD107a among tumor-associated NK cells. 6–11 mice per group from two independent experiments. *p < .05. (b) Mice were NK-depleted by i.p. injection of anti-NK1.1 antibody 24h before sham surgery (n = 9) or CLP (n = 12). Injection of anti-NK1.1 antibody was repeated once three days later. Tumor volumes were measured before (D14) and 7 days after surgery (D14 + 7). (c) Mice were NK-depleted by i.p. injection of anti-NK1.1 antibody 24h before PBS (n = 11) or LPS (n = 11) i.p. challenge. Injection of anti-NK1.1 antibody was repeated once three days later. Tumor volumes were measured before (D14) and 7 days after surgery (D14 + 7).
Sepsis promotes the anti-tumoral functions of NK cells
We sought to address whether a septic insult induced by either CLP or LPS may drive the anti-tumoral effector functions of NK cells. Splenic NK cells obtained from CLP-operated or LPS-challenged mice, or from their respective sham-operated or PBS-injected control counterparts, were purified and co-cultured with MHC-1low MCA205 target cells. We assessed the IFNγ-producing and cytotoxic capacities of NK cells as described above.
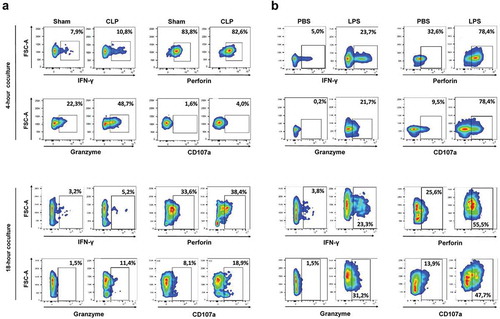
Splenic NK cells obtained from CLP-operated animals displayed a low-activation pattern, as assessed by low levels of intracellular and surface CD107a expressions (Figure S9). When put in the presence of MCA205 cells, they exhibited increased production of IFNγ and increased cytotoxic functions ()). Splenic NK cells obtained from LPS-challenged mice displayed a pre-activated pattern with increased expressions of IFNγ, perforin and granzyme B, but retained low CD107a expression (Figure S9). When co-cultured with MCA205 cells, they exhibited increased production of IFNγ, and also potent cytotoxic functions as suggested by sustained perforin and granzyme B expressions and increased CD107a expression consistent with an active degranulation process ()). Similar results were obtained using YAC-1 as target cells (Figure S10)
Figure 6. Sepsis enhances the NK cytotoxic functions towards MCA205 target cells. Mice were first s.c. inoculated with MCA205 cells, and then subjected to CLP or sham surgery (a), or to LPS or PBS challenge (b) 14 days after. Splenic NK cells pooled from 3 mice were purified at 24 hours following septic challenge, and co-cultured for 4 h and 18 h with MCA205 cells (effector-to-target ratio 1:1). Among NK cells, the intracellular expressions of IFNγ, perforin, granzyme B and the outer membrane expression of CD107a were quantified by flow cytometry. The figure is representative of two independent experiments.
Discussion
Infections and malignancies sustain a complex crosstalk and are likely to impact each other’s pathophysiology and course. Some bacterial, viral or parasitic pathogens are well known to promote cell transformation and carcinogenesis through genomic integration, chronic inflammation or sustained immune stimulation, and as such represent major health issues worldwide.Citation16 Whereas acute bacterial infections are common complications in cancer patients, few studies have investigated the impact of sepsis-induced immune dysfunctions on the development of oncological disorders. Some recent epidemiological data suggest that patients who survived an episode of bacterial sepsis are prone to further development of malignancies.Citation17–Citation19 Accordingly, sequential sepsis-then-cancer models have shown that post-septic mice inoculated with tumor cell lines exhibited accelerated tumor growth, related to the accumulation of tumor-associated granulocytic MDSC,Citation11 regulatory T cellsCitation6 or macrophages.Citation7 In contrast, the present reverse cancer-then-sepsis model is associated with both local and metastatic tumor growth inhibition. Altogether, our data show that sepsis harbors opposite effects on malignant tumor growth depending on the experimental sequence. We assume that such seemingly discrepant results may be related to prominent pro-inflammatory or immunosuppressive septic responses driving either inhibition or promotion of tumor growth, respectively.
Sepsis-induced inhibition in tumor growth echoes to various clinical and experimental observations. At the end of the 19th century, surgeon WB Coley had already reported “spontaneous” tumor regressions in patients with sarcoma who experienced acute streptococcal infections.Citation20 This observation led to the first attempts of immunotherapy in cancer patients who were treated with intra-tumoral inoculation with streptococcal preparations.Citation21 He thus demonstrated the potent efficacy of this so-called Coley’s toxin on various tumor types, including sarcoma, carcinoma, melanoma and lymphoma. Nowadays, several case reports have described unexpected oncological remissions in patients who survived severe bacterial infections.Citation10,Citation22–Citation24 Accordingly, experimental models have shown that acute infectious challenges applied concomitant to or after tumor cell inoculation were able to suppress tumor growth. Such antineoplastic properties have thus been attributed to various bacterial, viral and parasitic pathogens, including Proteus mirabilis, Chlamydia pneumoniae, Listeria monocytogenes, lymphocytic choriomeningitis virus, Toxoplasma gondii or Plasmodium yoelii, most of them being intracellular and thereby eliciting potent Th1 cell-mediated immune responses.Citation8–Citation10,Citation25,Citation26 In a mouse model of breast cancer, administration of virulence-attenuated Salmonella typhimurium potentiated the efficacy of anthracycline chemotherapy.Citation27 Commensal pathogens from the gut microbiota may also modulate the tumor microenvironment and prime immune cells towards enhanced anti-tumoral activity elicited by cytostatic chemotherapy or checkpoint inhibitors.Citation28–Citation30 Disruption in the gut flora by antibiotics may then impair the response to immune checkpoint inhibitors.Citation31
The models of CLP and LPS exhibit significant differences in the kinetics and magnitude of the inflammatory response.Citation32 A LPS challenge induces an early and potent but transient release of cytokines and chemokines, whereas it is more progressive and sustained following CLP. Inhibition in tumor growth was more pronounced following LPS challenge, in association with enhanced early degranulation by intra-tumoral NK cells. However, subtotal NK depletion could not restore tumor growth in LPS-challenged mice, suggesting that preserved cytotoxic activity of residual NK cells or enhanced tumoricidal activity of alternative cells such as macrophages may sustain potent antitumoral functions in this setting.Citation33,Citation34 Polymicrobial peritonitis induced by CLP involves both Gram-negative and Gram-positive bacteria that signal through various pathogen recognition receptors. Our results point to an immunoregulatory role of Tlr4 in sepsis-induced tumor inhibition. However, the impact of TLR-dependent signaling on tumor growth is equivoqual, depending on the respective results of TLR stimulation in tumor cells and their immune microenvironment.Citation35 TLR-expressing tumor cell lines may exhibit accelerated tumor growth and promote invasiveness when cultured in vitro in the presence of the corresponding TLR ligands.Citation36–Citation38 In transplanted mouse models, administration of PAMPs or functional modulation of TLR signaling may either exert a potent anti-tumoral effect or may conversely promote tumorogenesis and metastatic spread, depending on the experimental model, the tumor cell line and the stage of tumor development.Citation38–Citation43 Understanding how TLRs may shape anti-tumoral immune responses is paramount importance to improve the efficacy of the emerging strategies of immunotherapy and vaccines for the treatment of cancer.Citation44
The reverse sepsis-then-cancer and cancer-then sepsis sequential models resulted in opposite effects on tumor growth, and also differentially shaped intra-tumoral immune infiltration. In our initial sepsis-then-cancer model, CD11b+Ly6G+ granulocytic MDSC accounted for the largest proportion of tumor-infiltrating immune cells in post-septic animals and to a lesser extent in sham-operated ones.Citation11 In contrast, monocytes/macrophages accounted for the majority of tumor-infiltrating immune cells in the present cancer-then-sepsis model when tumor cells were inoculated to naive mice. However we did not observe any firm impact of sepsis on the functional modulation of tumor-associated monocytes/macrophages (data not shown), suggesting that alternative cell subsets were involved into the anti-tumoral activity elicited by sepsis.
Cytotoxic CD8+ T lymphocytes and NK cells are considered as the main tumoricidal cells. CD8+ T lymphocytes induce antigen-dependent cytotoxicity through the recognition of tumoral peptides by T-cell receptors. However, we did not observe any impact of sepsis on the effector functions of CD8+ T lymphocytes yet sensitized beforehand with tumor cells. NK cells are innate immune effectors able to distinguish tumor cells form healthy cells without prior sensitization. Target cells expressing low levels of MHC class I molecules and increased levels of activating ligands may then engage the cytotoxic activity of NK cells. In addition, the invariant NKT subset may become activated following the recognition of CD1d expressed by tumor cells themselves or by immune cells, and thereby promote tumor cell elimination, directly or indirectly through activation of other NK cells.Citation45 Granzyme A and granzyme B are mainly expressed by NK cells during bacterial sepsis and are involved in the regulation of inflammatory response and anti-bacterial defense.Citation46,Citation47 We focused on the role of granzyme B viewed as a key cytotoxic molecule for tumor cell death, whereas granzyme A is considered as quite dispensable in cancer immunosurveillance. In addition, NK cells are major sources of pro-inflammatory cytokines such as IFNγ and TNFα, thereby promoting the activation and maturation of dendritic cells, macrophages and T-cell lymphocytes. Hence, numerous studies have emphasized the critical role of NK cells in tumor control.Citation48 A low cytotoxic activity of NK cells is associated with an increased risk of developing cancer, and high numbers of tumor-infiltrating cytotoxic NK cells correlate with an improved prognosis in cancer patients. In experimental cancer models, depletion of NK cells or deficiency in NKG2D or NKp46 activation receptors enhances tumor growth. Expression of NKG2D ligands on tumor cells may also promote the activation of tumor-associated NK cells.Citation49,Citation50 In the present study, it is noteworthy that spleen and tumor-infiltrating NK cells displayed markedly different functional patterns, suggesting that the tumor microenvironment and the presence of target cells promoted NK cell activation regardless of the superimposed septic insult. The functional competence of NK cells may then result from the integration of both local and systemic signals. On one hand, tumor cells or their immune microenvironment deliver inhibitory signals likely to subvert the functions of NK cells.Citation51 On the other, sepsis-induced cytokine release may directly or indirectly increase the functions of NK cells.Citation52 Various cytokines such as IL-12, IL-15 and IL-18 can elicit IFNγ secretion by NK cells. Especially, the role of IL-12/IFNγ axis has been fully demonstrated in the immune interplay towards tumor rejection.Citation53–Citation55 Dendritic cells, already known as important coordinators of anti-infectious immune responses in sepsis, may also prime tumor-associated NK cells through IL15-mediated mechanisms to recover functional competence.Citation56,Citation57
Conclusion
Confrontation of the present study with our previous oneCitation11 provides firm evidence that sepsis harbors dual effects on malignant tumor growth depending on the sequence of insults. We herein demonstrated that sepsis applied to mice with cancer inhibited tumor growth in a Tlr4-dependent manner that was associated with enhanced anti-tumoral functions of NK cells. These findings may renew interest in anticancer bacterial immunotherapy, currently restricted to intravesical instillation of Mycobacterium bovis Calmette-Guérin bacillus for the treatment of bladder cancer.Citation58 Finally, this raises the question of the impact of sepsis on response to cytostatic chemotherapy or immunotherapy in critically ill cancer patients and their eventual long-term oncological prognosis.
Authors’ contributions
F Pène designed the study.
C Vigneron, A Mirouse, H Merdji, C Rousseau, C Cousin and F Alby-Laurent performed the research.
C Vigneron and F Pène drafted the manuscript.
C Vigneron, A Mirouse, H Merdji, C Rousseau, C Cousin, F Alby-Laurent, JP Mira, JD Chiche, JF Llitjos and F Pène analysed and interpreted the data, and contributed to writing the manuscript.
Availability of data and material
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable
Disclosure of potential conflicts of interest
The authors declare that they have no competing interests
Ethics approval
The study was approved by the Paris Descartes University ethics committee for animal experimentation, Paris, France (#12-094 & #17-054)
Meeting presentation
Presented in part at the 45th and 46th Congresses of the Société de Réanimation de Langue Française, Paris, France.
Supplemental Material
Download MS Word (2.7 MB)Acknowledgments
We are indebted to Matthieu Benard at the animal facility and to Patrice Vallin at the cytometry and immunobiology (CYBIO) facility of the Cochin Institute.
Supplemantary material
Supplemental data for this article can be access on the publisher’s website.
Additional information
Funding
References
- Zuber B, Tran TC, Aegerter P, Grimaldi D, Charpentier J, Guidet B, Mira JP, Pène F. Impact of case volume on survival of septic shock in patients with malignancies. Crit Care Med. 2012;40:55–62. doi:10.1097/CCM.0b013e31822d74ba.
- Venet F, Monneret G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol. 2018;14:121–137. doi:10.1038/nrneph.2017.165.
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi:10.1016/j.cell.2010.01.025.
- Hotchkiss RS, Moldawer LL. Parallels between cancer and infectious disease. N Engl J Med. 2014;371:380–383. doi:10.1056/NEJMcibr1404664.
- Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. doi:10.1038/nri.2016.107.
- Cavassani KA, Carson W, Moreira AP, Wen H, Schaller MA, Ishii M, Lindell DM, Dou Y, Lukacs NW, Keshamouni VG. The post sepsis-induced expansion and enhanced function of regulatory T cells create an environment to potentiate tumor growth. Blood. 2010;115:4403–4411. doi:10.1182/blood-2009-09-241083.
- Mota JM, Leite CA, Souza LE, Melo PH, Nascimento DC, de-Deus-Wagatsuma VM, Temporal J, Figueiredo F, Noushmehr H, Alves-Filho JC. Post-sepsis state induces tumor-associated macrophage accumulation through CXCR4/CXCL12 and favors tumor progression in mice. Cancer Immunol Res. 2016;4:312–322. doi:10.1158/2326-6066.CIR-15-0170.
- Chen L, He Z, Qin L, Li Q, Shi X, Zhao S, Chen L, Zhong N, Chen X. Antitumor effect of malaria parasite infection in a murine Lewis lung cancer model through induction of innate and adaptive immunity. PLoS One. 2011;6:e24407. doi:10.1371/journal.pone.0024407.
- Zhang H, Diao H, Jia L, Yuan Y, Thamm DH, Wang H, Jin Y, Pei S, Zhou B, Yu F. Proteus mirabilis inhibits cancer growth and pulmonary metastasis in a mouse breast cancer model. PLoS One. 2017;12:e0188960. doi:10.1371/journal.pone.0188960.
- Buzas K, Marton A, Vizler C, Gyukity-Sebestyen E, Harmati M, Nagy K, Zvara A, Katona RL, Tubak V, Endresz V, et al. Bacterial sepsis increases survival in metastatic melanoma: chlamydophila pneumoniae induces macrophage polarization and tumor regression. J Invest Dermatol. 2016;136:862–865. doi:10.1016/j.jid.2015.12.032.
- Llitjos JF, Auffray C, Alby-Laurent F, Rousseau C, Merdji H, Bonilla N, Toubiana J, Belaidouni N, Mira JP, Lucas B, et al. Sepsis-induced expansion of granulocytic myeloid-derived suppressor cells promotes tumour growth through Toll-like receptor 4. J Pathol. 2016;239:473–483. doi:10.1002/path.4744.
- Shu SY, Rosenberg SA. Adoptive immunotherapy of newly induced murine sarcomas. Cancer Res. 1985;45:1657–1662.
- International Cell Line Authentification Committee [Internet]. https://iclac.org/databases/cross-contaminations/.[accessed 2019 Feb 4]
- National Center for Biotechnology Information, BioSample database. http://www.ncbi.nlm.nih.gov/biosample/?term=cell%20line%20status%20misidentified[Attribute]. [accessed 2019 Feb 4].
- Pène F, Zuber B, Courtine E, Rousseau C, Ouaaz F, Toubiana J, Tazi A, Mira JP, Chiche JD. Dendritic cells modulate lung response to Pseudomonas aeruginosa in a murine model of sepsis-induced immune dysfunction. J Immunol. 2008;181:8513–8520. doi:10.4049/jimmunol.181.12.8513.
- de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, Plummer M. Global. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 2012;13:607–615. doi:10.1016/S1470-2045(12)70137-7.
- Kristinsson SY, Bjorkholm M, Hultcrantz M, Derolf AR, Landgren O, Goldin LR. Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. J Clin Oncol. 2011;29:2897–2903. doi:10.1200/JCO.2011.34.8540.
- Titmarsh GJ, McMullin MF, McShane CM, Clarke M, Engels EA, Anderson LA. Community-acquired infections and their association with myeloid malignancies. Cancer Epidemiol. 2014;38:56–61. doi:10.1016/j.canep.2013.10.009.
- Liu Z, Mahale P, Engels EA. Sepsis and risk of cancer among elderly adults in the United States. Clin Infect Dis. 2018.
- Coley WB II. Contribution to the knowledge of sarcoma. Ann Surg. 1891;14:199–220. doi:10.1097/00000658-189112000-00015.
- Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas: with a report of ten original cases. Am J Med Sci. 1893;105:487–511. doi:10.1097/00000441-189305000-00001.
- Ifrah N, James JM, Viguie F, Marie JP, Zittoun R. Spontaneous remission in adult acute leukemia. Cancer. 1985;56:1187–1190. doi:10.1002/1097-0142(19850901)56:5<1187::aid-cncr2820560536>3.0.co;2-o.
- Maywald O, Buchheidt D, Bergmann J, Schoch C, Ludwig WD, Reiter A, Hastka J, Lengfelder E, Hehlmann R. Spontaneous remission in adult acute myeloid leukemia in association with systemic bacterial infection-case report and review of the literature. Ann Hematol. 2004;83:189–194. doi:10.1007/s00277-003-0741-y.
- Kostner AH, Johansen RF, Schmidt H, Molle I. Regression in cancer following fever and acute infection. Acta Oncol. 2013;52:455–457. doi:10.3109/0284186X.2012.711954.
- Rankin EB, Yu D, Jiang J, Shen H, Pearce EJ, Goldschmidt MH, Levy DE, Golovkina TV, Hunter CA, Thomas-Tikhonenko A. An essential role of Th1 responses and interferon gamma in infection-mediated suppression of neoplastic growth. Cancer Biol Ther. 2003;2:687–693.
- Hunter CA, Yu D, Gee M, Ngo CV, Sevignani C, Goldschmidt M, Golovkina TV, Evans S, Lee WF, Thomas-Tikhonenko A. Cutting edge: systemic inhibition of angiogenesis underlies resistance to tumors during acute toxoplasmosis. J Immunol. 2001;166:5878–5881. doi:10.4049/jimmunol.166.10.5878.
- Saltzman D, Augustin L, Leonard A, Mertensotto M, Schottel J. Low dose chemotherapy combined with attenuated Salmonella decreases tumor burden and is less toxic than high dose chemotherapy in an autochthonous murine model of breast cancer. Surgery. 2018;163:509–514. doi:10.1016/j.surg.2017.09.036.
- Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342:967–970. doi:10.1126/science.1240527.
- Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342:971–976. doi:10.1126/science.1240537.
- Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CP, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079–1084. doi:10.1126/science.aad1329.
- Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2017;359:91–97. doi:10.1126/science.aan3706.
- Remick DG, Newcomb DE, Bolgos GL, Call DR. Comparison of the mortality and inflammatory response of two models of sepsis: lipopolysaccharide vs. cecal ligation and puncture. Shock. 2000;13:110–116.
- Prakash H, Nadella V, Singh S, Schmitz-Winnenthal H. CD14/TLR4 priming potentially recalibrates and exerts anti-tumor efficacy in tumor associated macrophages in a mouse model of pancreatic carcinoma. Sci Rep. 2016;6:31490. doi:10.1038/srep31490.
- Muller E, Christopoulos PF, Halder S, Lunde A, Beraki K, Speth M, Oynebraten I, Corthay A. Toll-like receptor ligands and interferon-gamma synergize for induction of antitumor m1 macrophages. Front Immunol. 2017;8:1383. doi:10.3389/fimmu.2017.01383.
- Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer. 2009;9:57–63. doi:10.1038/nrc2541.
- Huang B, Zhao J, Shen S, Li H, He KL, Shen GX, Mayer L, Unkeless J, Li D, Yuan Y, et al. Listeria monocytogenes promotes tumor growth via tumor cell toll-like receptor 2 signaling. Cancer Res. 2007;67:4346–4352. doi:10.1158/0008-5472.CAN-06-4067.
- Bohnhorst J, Rasmussen T, Moen SH, Flottum M, Knudsen L, Borset M, Espevik T, Sundan A. Toll-like receptors mediate proliferation and survival of multiple myeloma cells. Leukemia. 2006;20:1138–1144. doi:10.1038/sj.leu.2404225.
- Yang H, Wang B, Wang T, Xu L, He C, Wen H, Yan J, Su H, Zhu X. et al. Toll-like receptor 4 prompts human breast cancer cells invasiveness via lipopolysaccharide stimulation and is overexpressed in patients with lymph node metastasis. PLoS One. 2014;9:e109980. doi:10.1371/journal.pone.0109980.
- Pidgeon GP, Harmey JH, Kay E, Da Costa M, Redmond HP, Bouchier-Hayes DJ. The role of endotoxin/lipopolysaccharide in surgically induced tumour growth in a murine model of metastatic disease. Br J Cancer. 1999;81:1311–1317. doi:10.1038/sj.bjc.6694369.
- Harmey JH, Bucana CD, Lu W, Byrne AM, McDonnell S, Lynch C, Bouchier-Hayes D, Dong Z. Lipopolysaccharide-induced metastatic growth is associated with increased angiogenesis, vascular permeability and tumor cell invasion. Int J Cancer. 2002;101:415–422. doi:10.1002/ijc.10632.
- Standiford TJ, Kuick R, Bhan U, Chen J, Newstead M, Keshamouni VG. TGF-beta-induced IRAK-M expression in tumor-associated macrophages regulates lung tumor growth. Oncogene. 2011;30:2475–2484. doi:10.1038/onc.2010.619.
- Sfondrini L, Rossini A, Besusso D, Merlo A, Tagliabue E, Menard S, Balsari A. Antitumor activity of the TLR-5 ligand flagellin in mouse models of cancer. J Immunol. 2006;176:6624–6630. doi:10.4049/jimmunol.176.11.6624.
- Chang LS, Leng CH, Yeh YC, Wu CC, Chen HW, Huang HM, Liu SJ. Toll-like receptor 9 agonist enhances anti-tumor immunity and inhibits tumor-associated immunosuppressive cells numbers in a mouse cervical cancer model following recombinant lipoprotein therapy. Mol Cancer. 2014;13:60. doi:10.1186/1476-4598-13-60.
- Dajon M, Iribarren K, Cremer I. Toll-like receptor stimulation in cancer: A pro- and anti-tumor double-edged sword. Immunobiology. 2017;222:89–100. doi:10.1016/j.imbio.2016.06.009.
- Crowe NY, Smyth MJ, Godfrey DI. A critical role for natural killer T cells in immunosurveillance of methylcholanthrene-induced sarcomas. J Exp Med. 2002;196:119–127. doi:10.1084/jem.20020092.
- Arias MA, Jimenez de Bagues MP, Aguilo N, Menao S, Hervas-Stubbs S, de Martino A, Alcaraz A, Simon MM, Froelich CJ, Pardo J. Elucidating sources and roles of granzymes A and B during bacterial infection and sepsis. Cell Rep. 2014;8:420–429. doi:10.1016/j.celrep.2014.06.012.
- Garcia-Laorden MI, Stroo I, Blok DC, Florquin S, Medema JP, de Vos AF, van der Poll T. Granzymes A and B regulate the local inflammatory response during klebsiella pneumoniae pneumonia. J Innate Immun. 2016;8:258–268. doi:10.1159/000443401.
- Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. 2018;18:671–688. doi:10.1038/s41577-018-0061-z.
- Cheney EE, Wise EL, Bui JD, Schreiber RD, Carayannopoulos LN, Spitzer D, Zafirova B, Polic B, Shaw AS, Markiewicz MA. A dual function of NKG2D ligands in NK-cell activation. Eur J Immunol. 2012;42:2452–2458. doi:10.1002/eji.201141849.
- Deng W, Gowen BG, Zhang L, Wang L, Lau S, Iannello A, Xu J, Rovis TL, Xiong N, Raulet DH. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science. 2015;348:136–139. doi:10.1126/science.1258867.
- Hasmim M, Messai Y, Ziani L, Thiery J, Bouhris JH, Noman MZ, Chouaib S. Critical role of tumor microenvironment in shaping NK cell functions: implication of hypoxic stress. Front Immunol. 2015;6:482. doi:10.3389/fimmu.2015.00482.
- Guo Y, Patil NK, Luan L, Bohannon JK, Sherwood ER. The biology of natural killer cells during sepsis. Immunology. 2018;153:190–202. doi:10.1111/imm.12854.
- Lee NC, Tsung K, Norton JA. Production of interferon-gamma by tumor-sensitized T cells is essential for interleukin-12-induced complete tumor eradication. Surgery. 2002;132:365–368.
- Segal JG, Lee NC, Tsung YL, Norton JA, Tsung K. The role of IFN-gamma in rejection of established tumors by IL-12: source of production and target. Cancer Res. 2002;62:4696–4703.
- Li Z, Pradera F, Kammertoens T, Li B, Liu S, Qin Z. Cross-talk between T cells and innate immune cells is crucial for IFN-gamma-dependent tumor rejection. J Immunol. 2007;179:1568–1576. doi:10.4049/jimmunol.179.3.1568.
- Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity. 2007;26:503–517. doi:10.1016/j.immuni.2007.03.006.
- Guo Y, Luan L, Patil NK, Wang J, Bohannon JK, Rabacal W, Fensterheim BA, Hernandez A, Sherwood ER. IL-15 enables septic shock by maintaining NK cell integrity and function. J Immunol. 2017;198:1320–1333. doi:10.4049/jimmunol.1601486.
- Kramer MG, Masner M, Ferreira FA, Hoffman RM. Bacterial therapy of cancer: promises, limitations, and insights for future directions. Front Microbiol. 2018;9:16. doi:10.3389/fmicb.2018.00016.