
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The combination of immune-stimulating strategies has the potency to improve immunotherapy of cancer. Vaccination against neoepitopes derived from patient tumor material can generate tumor-specific T cell immunity, which could reinforce the efficacy of checkpoint inhibitor therapies such as anti-PD-1 treatment. DNA vaccination is a versatile platform that allows the inclusion of multiple neoantigen-coding sequences in a single formulation and therefore represents an ideal platform for neoantigen vaccination. We developed an anti-tumor vaccine based on a synthetic DNA vector designed to contain multiple cancer-specific epitopes in tandem. The DNA vector encoded a fusion gene consisting of three neoepitopes derived from the mouse colorectal tumor MC38 and their natural flanking sequences as 40 amino acid stretches. In addition, we incorporated as reporter epitopes the helper and CTL epitope sequences of ovalbumin. The poly-neoantigen DNA vaccine elicited T cell responses to all three neoantigens and induced functional CD8 and CD4 T cell responses to the reporter antigen ovalbumin after intradermal injection in mice. The DNA vaccine was effective in preventing outgrowth of B16 melanoma expressing ovalbumin in a prophylactic setting. Moreover, the combination of therapeutic DNA vaccination and anti-PD-1 treatment was synergistic in controlling MC38 tumor growth whereas individual treatments did not succeed. These data demonstrate the potential of DNA vaccination to target multiple neoepitopes in a single formulation and highlight the cooperation between vaccine-based and checkpoint blockade immunotherapies for the successful eradication of established tumors.
Introduction
Tumor cells accumulate somatic point mutations that can alter wild-type protein sequences making them immunologically different from healthy cells. T cells can detect these alterations by virtue of recognition of MHC-presented peptides up to single amino acid modifications, named antigenic neoepitopes.Citation1 However, the initiation of spontaneous tumor-specific T cell responses is limited by the lack of proper immune stimulation and is often dampened by the immune suppressive activity exerted by the tumor.Citation2 Checkpoint inhibitor therapies unleash these T cells and result in the long-term survival of patients with previously untreatable cancers.Citation3 Still, only a minority of patients benefits from checkpoint inhibitor therapies, leaving room for complementary strategies.Citation4 These may include vaccination against neoantigens, which can not only boost pre-existing responses but also induce de novo priming of tumor-specific T cells.
The design of personalized cancer vaccines harboring tumor mutations is still in early stage and needs to meet several requirements.Citation5 Exact prediction of the neoantigens likely to generate a peptide epitope that will bind to the relevant MHC alleles and induce functional T cell responses is still not fully achievable by the current in silico systems used for epitope prediction. Therefore, it is required to include a sufficient number of candidate sequences to increase the chances of including actual T cell epitopes in the vaccine. Furthermore, the inclusion of multiple antigens could promote the generation of a broad immune response, which may enhance vaccine efficacy and contribute to counteract immune suppression. Another requisite for patient-tailored cancer vaccines is versatility in synthesis and production of several different sequences, as the heterogeneous array of antigen sequences varies across individual patients. This aspect is not trivial in classical peptide-based systems, as amino acid sequence dictates the physicochemical properties of the vaccine, adding complications to the manufacturing process and formulation.Citation6 In short, an ideal neoantigen vaccine platform should be flexible enough to be able to incorporate a multitude of epitopes and allow fast and reliable production independently of the exact amino acid sequences of the selected epitopes.
In the last few years, efforts in refining neoantigen identification and formulation of cancer vaccines for therapeutic treatment have demonstrated the potential of this approach in preclinical models for synthetic peptide- and RNA-based vaccines.Citation7-Citation10 These studies have led the way for the first in-human application in two independent pioneering trials in melanoma patients.Citation11,Citation12 Vaccination with neoepitopes derived from single amino acid mutations selected upon sequencing of patients’ material elicited tumor-specific T cell responses with clinical benefits both with peptide- and RNA-based vaccines.
Up until recently, DNA-based vaccines targeting neoantigen have been scarcely explored. DNA represents a versatile platform that can accommodate any sequence without affecting its stability or solubility. In addition, DNA is easily synthesized and production costs are relatively low. DNA vaccines were first shown to be immunogenic nearly 30 years ago.Citation13-Citation15 Since then, numerous studies have explored the potential of gene immunization. Methods for optimizing administration routes, delivery, and plasmid design have been central in a variety of preclinical and clinical studies.Citation16 Several studies demonstrated that immune responses can be induced by intramuscular, intradermal or intravenous administration of DNACitation14,Citation17,Citation18 and original administration devices such as gene gun,Citation19 electroporationCitation20 and tattooingCitation21 have been employed to improve transfection efficiency and induction of both humoral and cellular immune responses. A recent study using electroporation-mediated DNA delivery of multiple neoantigen constructs showed effective induction of anti-tumor CD8 T cell responses in mice.Citation22
In this study, we show the efficacy of DNA vectors as a vaccine carrier for multiple neoantigens based on a string-of-bead design. Using regular intradermal injection without the need for specialized equipment or an adjuvant, the DNA vaccine induced multiple CD8 and CD4 T cell responses against both reporter epitopes and neoantigens. We demonstrate that vaccination enables T cell-mediated anti-tumor control in a prophylactic as well as in a therapeutic setting. Furthermore, we show that DNA vaccination can synergize with and improve the efficacy of checkpoint inhibitor therapy.
Methods
Animals: For vaccination and tumor experiments, 6–8 weeks old female C57BL/6 were purchased from Charles River Laboratories. The TCR transgenic OT-I and OT-II mouse strains were obtained from Jackson Laboratory and maintained on CD45.1+ C57BL/6 background. Mice were housed in specific pathogen-free (SPF) conditions at the LUMC animal facility. All animal experimentations were approved by and according to guidelines of the Dutch Animal Ethical Committee.
Cell lines: The B3Z hybridoma cell line was cultured in IMDM medium (Lonza) supplemented with 8% FCS (Greiner), penicillin and streptomycin, glutamine (Gibco), β-mercaptoethanol (Merck), hygromycin B (AG Scientific Inc) to maintain expression of the beta-galactosidase reporter gene. B16-OVA and MC38 tumor cell lines were cultured in IMDM medium supplemented with penicillin/streptomycin, glutamine, and 8% FCS. B16-OVA were cultured in the presence of G418 (Life Technologies) for maintenance of OVA expression.Citation23 For ex vivo stimulation of lymphocytes, the dendritic cell line D1 was used and cultured as previously described.Citation24
DNA construct and peptides: Codon-optimized antigen sequences, fuzed by alanine linkers were synthesized and cloned into a CMV-driven expression vector containing a rabbit beta-globin poly-A signal and kanamycin resistance marker (ATUM). As control, DasherGFP was cloned in the same plasmid vector. Plasmids were propagated in E. coli cultures and purified using Nucleobond Xtra maxi EF columns (Macherey-Nagel) according to manufacturer’s instructions. For vaccination, plasmids were column-purified twice, each time using a fresh column, and dissolved at 3 mg/ml in TrisEDTA buffer (1:0.1 mM).
Synthetic long peptides for the five epitopes were synthetized by LUMC peptide facility SIIVFNLLELEGDYR (Dpagt), LFRAAQLANDVVLQIM (Reps1), ELASMTNMELMSSIV (Adpgk), ISQAVHAAHAEINEAGR (OVA CD4), DEVSGLEQLESIINFEKLAAAAAK (OVA CD8) and used as peptide controls for all experiments.
In vitro transfection and antigen recognition assay: 3ʹ000 MC38 cells were seeded overnight in 96-well flat-bottom plates. Next day, cells were transfected using the SAINT-DNA transfection kit (SD-2001, kindly provided by Synvolux). In brief, a solution of plasmids and cationic lipids was mixed in a ratio 1:20 (µg DNA: µl Saint-DNA) in titrating quantities. SIINFEKL presentation by H2-Kb was detected with 25-D1.16 antibodyCitation25 in-house conjugated to Alexa 647. After 48 h, 50ʹ000 B3Z cells per well were added and incubated with transfected cells overnight. The following day, TCR activation triggered by recognition of the SIINFEKL epitope was detected by measurement of absorbance at 570 nm upon color conversion of chlorophenol red-β-D-galactopyranoside (Calbiochem®, Merck).
In vivo proliferation of adoptively transferred OT-I and OT-II cells: Naïve C57BL/6 mice received an intradermal injection of lipoplexes comprising vaccine or control plasmids complexed to cationic lipid SAINT18 (kindly provided by Synvolux)Citation26 7, 4, 2 or 0 days prior transfer of ovalbumin-specific OT-I and OT-II cells. CD8+ cells or CD4+ cells were isolated from spleens and lymph nodes of CD45.1+ OT-I or OT-II mice with enrichments sets (BD Biosciences), labeled with 5 µM CFSE (Invitrogen) and intravenously injected in vaccinated mice. Three days after transfer, proliferation of OT-I and OT-II cells was measured in lymph nodes and spleens by CFSE detection in CD45.1+/CD8+ or CD45.1+/CD4+ T cells.
Vaccination with peptide mix: Peptide vaccination was used as a positive control for priming and tumor experiments in vivo. It consisted of a mix of 50 µg of the five long peptides containing the five epitopes encoded in the DNA vaccine. The formulation was adjuvanted with 20 µg of poly(I:C) (Invivogen)
Priming of endogenous T cells: Naïve C57BL/6 mice were injected with plasmid-SAINT18 complexes in a 1:0.75 ratio (µg DNA: nmole SAINT18) in 0.9% NaClCitation26 either intradermally (30 µl), subcutaneously (30 µl), intramuscularly (30 µl), intraperitoneally (100 µl) or intravenously (100 µl) and boosted after 14 and 28 days. The level of SIINFEKL-specific CD8 T cells was monitored in blood with labeled tetramers. Twelve days after second booster injection, splenocytes were harvested and expanded for 1 week with D1 dendritic cells loaded with long peptide pools. Intracellular staining was performed upon stimulation with individual long peptides overnight in presence of 2 µg/ml Brefeldin A (Sigma Aldrich). T cells and cytokines were detected by antibody staining and analyzed with FlowJo software. The following antibody mix was used: eFluor450 anti-CD3, PE-Cy7 anti-CD4 (eBioscience), BV605 anti-mouse CD8α, APC anti-IFNγ (Biolegend), FITC anti-TNFα (eBioscience), PE IL-2 (eBioscience).
In vivo specific killing: Naïve C57BL/6 were primed and boosted after 21 days with 10 or 100 µg of plasmid-SAINT18 complexes. Twenty-one days after boost, vaccinated mice received peptide-loaded splenocytes to measure the cytotoxic activity of endogenously primed T cells. To this end, splenocytes were harvested from CD45.1+ or WT C57BL/6 naïve mice, labeled with 5, 0.25 or 0.0025 µM CFSE and differentially loaded for 1 h at 37ºC with 1µM SIINFEKL, Adpgk or Reps1 epitopes or an irrelevant peptide epitope derived from the E6 protein of Human Papilloma Virus (sequence: RAHYNIVTF). 4ʹ000ʹ000 splenocytes per peptide-loaded group were injected intravenously in vaccinated mice. One day after transfer, mice were sacrificed and single-cell suspension was analyzed by flow cytometry. Specific killing was calculated according to the following equation:
Prophylactic vaccination and B16-OVA tumor challenge: Naïve C57BL/6 female mice were vaccinated intradermally with 10 or 90 µg of plasmid-SAINT18 complexes. At day 42 (21 days after booster injection) 50ʹ000 B16-OVA cells were injected subcutaneously in the flank and tumor growth was monitored. Mice were sacrificed when the tumor volume surpassed 1000 mm3.
MC38 tumor challenge and therapeutic vaccination: Naïve C57BL/6 female mice were injected subcutaneously in the flank with 350ʹ000 MC38 cells and tumor growth was monitored. When tumors reached a palpable size with an estimated volume of 1 to 2 mm3 (day 5), mice were vaccinated with 10 µg of plasmid-SAINT18 complexes. Three and 7 days after vaccination, 50 µg of anti-PD-1 (Clone RMP1-14, InvivoPlus, BioXCell) antibody was injected subcutaneously next to the tumor mass.Citation27 Mice were sacrificed when the tumor volume surpassed 1ʹ000 mm3.
Statistical analysis: Results are expressed as mean ± SD. Statistical significance among groups was determined by multiple comparisons using the Graphpad software after ANOVA or non-parametric Kruskal–Wallis test. Cumulative survival time was calculated by the Kaplan–Meier method, and the log-rank test was applied to compare survival between two groups. P-values of ≤0.05 were considered statistically significant.
Results
Development of a poly-neoantigen DNA vaccine for in vitro and in vivo antigen presentation to T cells.
We aimed to include multiple antigenic sequences in a single DNA vaccine construct, and therefore we designed a plasmid encoding five epitopes in tandem in a single open reading frame ()). Three epitopes (Dpagt, Reps1, Adpgk) are described neoantigens containing specific point-mutated MHC class I binding sequences present in the mouse colon carcinoma cell line MC38.Citation7 The other two epitopes are, respectively, the helper (Help) and the cytotoxic T lymphocyte (CTL) epitopes from the model antigen chicken ovalbumin (OVA), and were included as control reporter epitopes for CD4 and CD8 T cell responses, respectively. Every epitope is flanked by its natural amino acid sequence for a total length of approximately 40 amino acids, and is linked to the next epitope by a linker encoding four alanines. Transcription is driven by the strong viral promoter of the immediate early gene 1 (IE1) of human cytomegalovirus (HCMV).
Figure 1. The poly-antigen DNA vaccine activates antigen-specific CD8 and CD4 T cells in vivo. (a) Schematic representation of the neoantigen DNA vaccine and the resulting poly-epitope peptide sequence. Direction of the open reading frame (ORF) is indicated. The individual CD8 and CD4 epitopes in the peptide are encircled in dark or light blue, respectively. For each of the three neoepitopes, the amino acid (aa) change resulting from somatic mutation is highlighted in red. (b) Upper panel: Quantification of the mean fluorescence intensity (MFI) using 25-D1.16 antibody of the SIINFEKL peptide presentation on MHC I molecule after transfecting MC38 cells in vitro with either the GFP plasmid (negative control) or the neoantigen plasmid. Lower panel: Activation of SIINFEKL-specific T cell hybridoma B3Z cells by MC38 cells transfected with the neoantigen DNA vaccine. The SIINFEKL synthetic peptide (1 µM) was added as a positive control (orange bars), and a plasmid coding for GFP was used as a negative control. Statistical significance was determined by one-way ANOVA followed by multiple comparison, *** p < 0.001 (c) Proliferation of adoptively transferred OT-I and OT-II cells, 3 days after intradermal injection of 10 µg of the neoantigen DNA vaccine. (d) Kinetics of in vivo antigen presentation to OT-I or OT-II cells after injection of the DNA construct. Proliferation of the OT-I (blue lines) and OT-II cells (red lines) measured by CFSE dilution in the inguinal lymph nodes of C57BL/6 mice immunized with either neoantigen DNA vaccine (left panel) or 50 µg of OVA CTL and helper peptides (right panel), used as positive controls for OT-I and OT-II cells proliferation, respectively. Error bars indicate mean ± SD, N = 2. (e) Proliferation of adoptively transferred OT-I and OT-II cells, 3 days after intradermal injection of 10 µg of different variants of neoantigen DNA vaccine. Error bars indicate mean ± SD, N = 2. Significance in relation to the GFP plasmid negative control was determined by one-way ANOVA followed by multiple comparisons. * p < 0.05, ** p < 0.01.
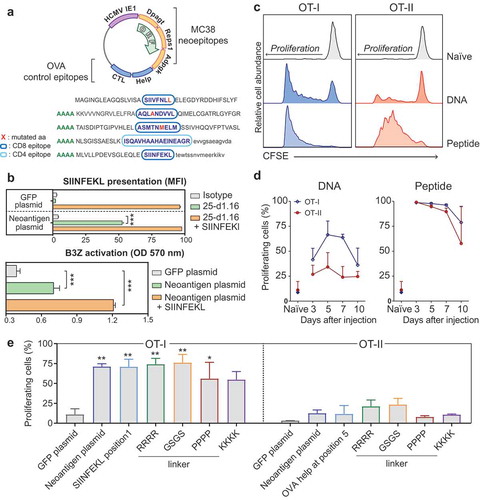
We first analyzed whether these five artificially connected sequences lead to the generation of the expected peptide epitopes and their presentation on MHC molecules. Upon transfection of the designed DNA construct, the translated protein product needs to be processed in such a way that the T cell epitopes are generated and presented by MHC molecules. MC38 cells, which do not express the ovalbumin gene, were transfected with the neoantigen construct and the presentation of the ovalbumin CTL epitope SIINFEKL was detected by staining the SIINFEKL/H2-Kb complex with the 25-D1.16 antibody (), upper panel). Transfection with the poly-neoantigen construct, but not with a control GFP-encoding construct, displayed positive staining for SIINFEKL/H2-Kb complexes. Moreover, after transfection with the neoantigen construct, cells were recognized by the hybridoma T cell line B3Z, which express a TCR specific for SIINFEKL/H2-Kb (), lower panel).
Next, we tested the ability of the neoantigen DNA construct to transfect cells and present the expected reporter ovalbumin epitopes in vivo. The plasmid was injected intradermally in mice 7, 4, 2, or 0 days prior transfer of CFSE-labeled OT-I and OT-II T cells, which possess transgenic TCRs specific, respectively, for the CTL and the helper epitopes of ovalbumin. Antigen induced proliferation of these cells was analyzed 3 days after transfer in draining lymph nodes and spleen (, Fig. S1 A and B). Injection of the construct was able to induce both OT-I and, to a lesser extent, OT-II proliferation, confirming successful transfection and presentation of the epitopes also in vivo. OT-I and OT-II proliferation upon DNA vaccine injection exhibited different kinetics compared to traditional synthetic peptide vaccine ()). DNA vaccination presents a slower onset of T cell proliferation compared to peptide vaccination, with optimal induction between 5 and 7 days after DNA vaccination (), left panel), as opposed to 3 days for peptide vaccination (), right panel). Finally, we evaluated whether the position of the epitopes or the artificial linker sequence between them could influence the efficiency of antigen presentation in vivo. To test this, we created a variant of the original neoantigen DNA construct in which the reporter SIINFEKL epitope was positioned as the first epitope at the N-terminal end of the polypeptide and additional variants in which the epitopes were connected via different amino acid linkers. We evaluated OT-I and OT-II proliferation upon vaccination in vivo ()). Overall, we observed no significant differences in OT-I or OT-II proliferation between the variants tested. Altogether these results demonstrate efficient MHC surface presentation of the CD8 and CD4 reporter epitopes of ovalbumin, irrespective of the position and linker sequence in the poly-antigen encoding DNA construct. This shows the feasibility of the string-of-bead design as a method to target multiple antigens in a single vaccine construct.
The DNA vaccine primes neoantigen-specific t cell responses in vivo
Next, we evaluated the ability of our DNA vaccine to generate all five encoded epitopes in vivo and its ability to induce de novo priming of antigen-specific T cells in wild-type C57BL/6 mice. Previous studies have highlighted an influence on vaccine efficacy depending on its formulation, methods, and routes of administration.Citation14,Citation17,Citation18,Citation28 To determine the optimal delivery route of our designed DNA vaccine, the construct was administered to mice via different routes (intradermal, subcutaneous, intramuscular, intraperitoneal and intravenous) and the immune response was boosted twice in intervals of 2 weeks (Fig. S2A). Tetramer staining in blood at several time points revealed effective priming of SIINFEKL-specific CD8 T cells for the groups that intradermally and intravenously received the DNA vaccines (Fig. S2B and S2C). In addition, splenocytes of vaccinated mice restimulated ex vivo with peptide-loaded dendritic cells displayed responses for all five epitopes encoded by the DNA vaccine (Fig. S2D and S2E). We concluded that intradermal injection of the neoantigen DNA vaccine was able to induce de novo priming of T cells upon classical needle-mediated administration in vivo.
To evaluate the cytotoxic function of the DNA vaccine-induced CD8 T cells, we analyzed the kinetics of the SIINFEKL-specific CD8 T cells and their capacity to specifically kill target cells presenting the epitopes. We also explored the dosing of plasmid administration to optimize the T cell response. C57BL/6 mice were vaccinated and boosted with two different doses of a DNA vector or with peptides, and the ability to kill splenocytes loaded with either SIINFEKL, Adpgk or Reps1 peptide was determined ()). The kinetics of DNA vaccination was slower compared to the synthetic peptide vaccination, as the peak of the priming response appeared 3 days later ()). After the boost, T cell responses to DNA and peptide vaccination were similar. Considering the dose, 100 µg of DNA appeared to be more effective mostly in the priming phase.
Figure 2. DNA vaccine primed T cells are functional. (a) Schematic representation of the vaccine administration, tetramer staining, target cells injection and specific killing analysis schedule in C57BL/6 mice. Mice were vaccinated intradermally with 10 or 100 µg of DNA (or a mix of peptides as positive control). SIINFEKL-specific responses were monitored in blood at different time points. To evaluate the killing capacity of the responses induced after vaccination, mice were injected with CFSE-labeled splenocytes loaded with minimal peptides and specific killing was analyzed two days later. (b) Kinetics of the SIINFEKL-specific CD8+ T cells responses induced by the vaccines, measured by SIINFEKL-H2-Kb tetramersand reported as percentage of total CD8+ T cells. (c). Representative flow cytometry histograms of CFSE-labeled antigen- or control peptide-loaded splenocytes detected in naïve and vaccinated mice. Two days after transfer, these target cells were detected in the spleen and specific killing was calculated. Percentages represent the relative proportions between cells loaded with an irrelevant control peptide (in gray) and target cells (white, orange, green or blue). (d) Specific killing by T cells in naïve mice versus mice vaccinated with DNA (10 or 100 µg) or peptide after transfer of antigen-loaded splenocytes. Error bars indicate mean ± SD.
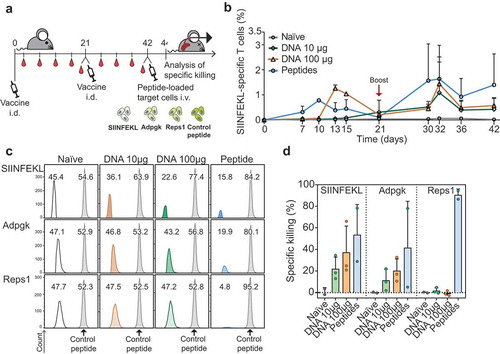
CD8 T cells primed by DNA were effective in killing SIINFEKL and Adpgk peptide-loaded T cells at day 44. Cytotoxicity against the Reps1 epitope was not detected upon DNA vaccination (,)). Vaccination with a higher dose of DNA marginally improved the killing capacity of the SIINFEKL-specific CD8 T cells and correlated with the levels of tetramer-specific cells present in blood two days before injecting target cells (see ), day 42). Based on these results we concluded that the poly-neoantigen DNA vaccine is able to induce functional CD8 T cells against multiple epitopes and we proceeded to evaluate its efficacy in immune control of cancer.
Prophylactic and therapeutic DNA vaccination elicits tumor control
We next investigated whether the T cell responses induced by DNA vaccination were able to provide immune control of tumors in vivo for both the OVA reporter epitopes and the neoantigens.
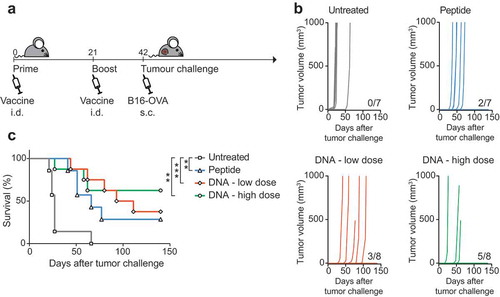
First, we evaluated anti-tumor efficacy for the reporter ovalbumin epitopes. Mice were prophylactically vaccinated with DNA or peptides before being challenged with the OVA-expressing melanoma cell line B16-OVA ()). To explore the impact of DNA dosing on the induction of T cell responses, two different amounts of DNA were tested and the induction of ovalbumin-specific CD8 responses was monitored by tetramer staining in blood samples (Fig. S3). Unvaccinated control mice developed tumors within 20 days from challenge. Mice vaccinated with DNA developed tumors later than unvaccinated controls, and a significant number of mice were fully protected from this aggressive tumor. A lower dose of vaccine corresponded to a lower protection but was still effective to prevent tumor growth in ~40% of the animals (,)). Vaccination with a higher dose of DNA resulted in full protection of 60% of the mice (,)). Hence, DNA vaccines were effective in inducing protective antitumor T cell responses, comparable to or better than the mice that received synthetic peptide vaccination. Moreover, a higher dose of vaccine corresponded to stronger protection, and this dose was used for further studies in a therapeutic setting.
Figure 3. DNA vaccination protects from challenge with B16-OVA. (a) Schematic representation of the schedule followed for vaccine administration and tumor challenge in C57BL/6 mice. Mice were vaccinated with a low (10 µg) or a high (90 µg) dose of DNA or with peptide and subsequently challenged with B16-OVA melanoma cells. Tumor growth was monitored for 150 days after challenge. (b) Tumor growth curves (represented in mm3) of individual mice in non-vaccinated versus vaccinated groups. The number of tumor-free mice for each vaccination group is indicated. Shown is one of two independently performed experiments which resulted in similar outcomes. (c) Overall survival of mice either untreated or vaccinated with peptide or DNA vaccines. Statistical significance was determined via Log-rank Mantel-Cox test. **p < 0.01, ***p < 0.0001.
After demonstrating the potential for antitumor activity of the DNA vaccine in the B16 melanoma model, which expresses the ovalbumin antigen but not the MC38-specific neoantigens, we evaluated the therapeutic efficacy of the same DNA vaccine in the MC38 tumor model. Mice were first inoculated with the MC38 colon carcinoma cell line, expressing the three neoantigens Dpagt1, Reps1 and Adpgk but not the ovalbumin epitopes. Mice with established tumors were vaccinated therapeutically on day 5 followed by a booster vaccination at day 26. As MC38 is known to exert a strong immunosuppressive effect,Citation29 we combined the vaccine with the immunomodulatory anti-PD-1 antibody treatment on day 8, 12, 22 and 29 ()). Without any treatment, tumors progressed rapidly and all mice succumbed within 21 days from tumor inoculation. Vaccination with DNA or peptides gave little or no delay and eventually all mice showed rapid tumor outgrowth, except for one mouse in the DNA vaccinated group. Anti-PD-1 treatment induced some delay in tumor growth but was not sufficient to prevent tumor outgrowth. Remarkably, when anti-PD-1 treatment was combined with DNA vaccination, tumor growth was significantly delayed and 25% of mice were able to clear the tumor and survive long term (,)). Notably, this effect was only observed with DNA vaccination but could not be achieved with the synthetic peptide vaccine.
Figure 4. Therapeutic DNA vaccination combined with anti-PD-1 treatment promotes tumor eradication in CD8 T cell-dependent manner. (a) Schematic representation of the MC38 tumor challenge experiment in C57BL/6 mice in therapeutic setting. Mice were injected subcutaneously with MC38 cells at day 0 and vaccinated with the DNA construct at days 5 and 26. Vaccination was combined with anti-PD-1 treatment at days 8, 12, 22 and 29. Tumor growth was monitored for 60 days after challenge. (b) Tumor growth curves (measured in mm3) of individual mice in untreated or treated with single or combined anti-PD-1, DNA vaccine and peptide mix. The number of tumor-free mice for each group is indicated. (c). Overall survival of mice represented in (b).(d). Schematic representation of the MC38 tumor challenge experiment in C57BL/6 mice and depletion of CD8 cells during vaccine and anti-PD-1 combination treatment. Mice were injected subcutaneously with MC38 cells at day 0, injected with anti-CD8 antibody at day 5, vaccinated with DNA construct and injected with anti-PD-1 antibody at days 5, 8 and 12. (e) Tumor growth curves (measured in mm3) of individual mice untreated or treated with single or combined anti-PD-1, DNA vaccine and anti-CD8. The number of tumor-free mice for each group is indicated. Vaccination with a control DNA construct (DNA-GFP) that does not contain the three MC38-specific neoantigens is used as a negative control. (f) Overall survival of mice represented in (e). Statistical significance was determined via Log-rank Mantel-Cox test. * p < 0.05, **p < 0.01, *** p < 0.001, **** p < 0.0001.
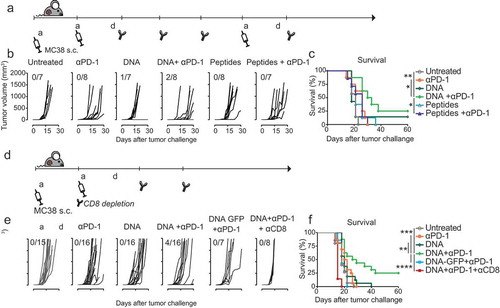
In addition, a single dose of the DNA vaccine combined with anti-PD-1 treatment ()) also resulted in a substantial delay of tumor outgrowth and complete tumor clearance was observed in some mice, resulting in a 25% cure rate. (,)). Importantly, this effect was not observed when vaccination was performed with a GFP-coding plasmid and was abolished when CD8 T cells were depleted right after vaccination, indicating the implication of neoantigen-specific CD8 T cell responses in tumor growth control. Altogether these data demonstrate that the designed poly-neoantigen DNA construct is an effective vaccine vector and that this design holds potential for neoantigen vaccination for specific immunotherapy of cancer.
Discussion
In this study, we demonstrate that a poly-neoantigen DNA vaccine not only provides prophylactic protection against tumor challenge but also synergizes with PD-1 blockade for tumor control in a therapeutic setting. The versatile DNA platform presented here allows the inclusion of multiple epitopes in tandem derived from multiple antigenic sequences, increasing the chances of triggering relevant T cell responses to improve the overall effectiveness of neoantigen-specific immunotherapy.
Our vaccine vector was able to induce functional responses without any additional adjuvant. DNA vectors may act as self-adjuvating vaccines as the innate immune system possesses various means to sense foreign or cytoplasmic DNA and activates an inflammatory response. Low-unmethylated CpG-rich regions linked to bacterial production of the DNA vaccine may contribute to immunogenicity via TLR9 signaling.Citation30,Citation31 Furthermore, cytoplasmic sensors in the STING axis were also reported to play a role in DNA-mediated immunization.Citation32 Nevertheless, efforts in optimizing the immunogenicity of DNA vaccines demonstrated that the inclusion in the sequence of pro-inflammatory cytokines such as IL-12Citation33 and GM-CSFCitation34-Citation36 and costimulatory-molecules such as B7-1, B7-2Citation37,Citation38 or CD40LCitation39 have a beneficial effect in generating effective immune responses. As we report significant but partial tumor control, it will be of interest to improve the cure rate by including such genetic adjuvants, which can readily be incorporated in the vaccine sequence.
The induction of CD8 as well as CD4 T cell responses is critical for cancer immunotherapy, as the ability of cytotoxic CD8 T cells to effectively attack and kill tumor cells depends on the presence of concomitant help provided by CD4 T cells.Citation40,Citation41 This is especially important given the high frequency of CD4 neoepitopes in tumor cells,Citation9,Citation11,Citation12 and given recent observations that CD4 T cells can control tumors independently of CD8 T cells.Citation42,Citation43 In a recent report, applying neoantigen DNA vaccination with electroporation resulted preferentially in the induction of CD8 responses.Citation22 In contrast, our intradermal DNA vaccination approach efficiently induced both CD8 and CD4 responses. Our data indicate that both MHC I and MHC II presentations occur; however induction of CD8 T cell responses appears more pronounced than induction of CD4 responses. This may suggest that antigen presentation is performed mainly by directly transfected cells and consequently cytosolic antigen is more efficiently presented. MHC II presentation occurs mainly on exogenously acquired antigen by specialized antigen-presenting cells (APCs). Previous reports investigating the working mechanism of intradermal DNA vaccination have highlighted that transfection takes place both in epidermal cells and, to a lesser extent, directly in professional APCs.Citation44,Citation45 It is still controversial whether antigen presentation upon DNA vaccination occurs by directly transfected cells or antigen is indirectly acquired from transfected cells by APCs.Citation46We believe that it will be important to elucidate the mechanism in the context of intradermal DNA vaccination in order to control and elicit optimal MHC II presentation.
The chosen colon cancer cell line MC38 tumor model represents a clinically relevant tumor both in light of neoantigen vaccination studies and immunomodulatory treatments. MC38 is known to induce spontaneous-CD8-mediated immune responses in mice with growing tumors, but, due to its highly immunosuppressive microenvironment, these T cells are apparently inactive and not able to eradicate tumor cells.Citation29 Treatment of MC38 tumor-bearing mice at early stages with immunomodulating antibodies against PD-1 or PD-L1, elicit effector T cell responses which can mediate tumor regression.Citation27 Here we show that vaccination against the selected neoantigensCitation7 in combination with anti-PD-1 can mediate tumor regression in a CD8 T cells-dependent fashion, while anti-PD-1 antibody by itself could not effectuate tumor clearance. Physiologically, the PD-1 axis contributes to negatively regulate peripheral-activated CD8 T cells, but malignant cells exploit this mechanism to shut down spontaneous tumor-specific T cells responses. Indeed, a recent report by Xiong and colleagues showed that neo-epitope specific CD8 T cells express a high level of co-inhibitory molecules, including PD-1.Citation47 When CD8 T cells are properly activated, by means of vaccination, for example, they also upregulate PD-1 and are therefore more susceptible to immune suppression. Accordingly, integration of anti-PD-1 blockade therapy resulted in complete response in both RNA- and peptide-based vaccination clinical studies for, respectively, oneCitation11 and twoCitation12 patients that experienced recurrence after vaccination. Altogether these observations show that specific immunotherapy can synergize with PD-1 checkpoint therapy most likely by supporting adequate effector functions of the increased frequencies of tumor-specific T cells.
Immunization with multiple epitopes in one formulation may result in reduced responses to individual epitopes. The occurrence of immune-dominant neoantigens has been reported in several studies.Citation48-Citation50 Nevertheless, a study identifying neoepitopes in patients with chronic leukemia reports how immune-dominance plays a role primarily in the induction of spontaneous responses, while vaccination against multiple epitopes diversifies the tumor-specific T cell repertoire and amplifies the heterogeneity of tumor-specific T cell responses.Citation50 In addition, tumor immunoediting could lead to antigen loss and the outgrowth of resistant tumor variants that do not possess one or more of the targeted neoantigens. The inclusion of multiple epitopes in a vaccine may be important to avoid the outgrowth of such resistant clones. Therefore, the beneficial effects of a more diversified T cell response are likely to outweigh a potential reduction of individual T cell specificities due to immuno-dominance.
Interestingly, we observed that primarily the Adpgk neoepitope appears to induce effector T cells which are able to recognize and eliminate antigen-loaded cells (see )). In contrast, DNA-induced Reps1-specific T cells were not able to kill target cells as opposed to the responses induced by peptide vaccination. Differences in induction of T cell responses depending on the method of immunization have also been reported in RNA vaccination studies.Citation9 Why these differences between peptide and gene immunization occur is as yet not clear; however, considering the notion that some responses to tumor neoantigens can still be irrelevant for tumor eradication, Citation51 these observations support the rationale of including multiple potential neoantigens in therapeutic cancer vaccines.
Personalized therapy against tumor neoantigens represents an exciting prospect for clinical translation. A personalized therapeutic cancer vaccine requires a flexible, cost-effective vaccine platform. Here, we show a proof of concept of a DNA vector as a versatile vaccine platform for the inclusion of multiple tumor neoantigens. Moreover, we show that this DNA vaccine synergizes with anti-PD-1 treatment in tumor control. Our data report the potency of stimulating tumor-specific responses via DNA vaccination in a string-of-bead design to achieve effective immunotherapy and underline the importance of combining different immunotherapy strategies in order to achieve effective clinical responses.
Authors’ contributions
ET, KO, MC, SvD, WH, JvB designed and performed experiments. ET, JvB, RA, GZ, and FO interpreted the experiments. ET and TA wrote the manuscript. All authors contributed to and approved the final manuscript.
Disclosure of potential conflicts of interest
KO, WH, JvB, and GZ are employees of Immunetune BV.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Supplemental Material
Download Zip (2.5 MB)Acknowledgments
The authors would like to thank Bram Teunisse and Mandy Los of Immunetune, Leiden, The Netherlands for their technical assistance and Eduard Talman of Synvolux, Leiden, The Netherlands for the production of SAINT18 cationic lipids.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Lurquin C, Van Pel A, Mariamé B, De Plaen E, Szikora JP, Janssens C, Reddehase MJ, Lejeune J, Boon T. Structure of the gene of tum- transplantation antigen P91A: the mutated exon encodes a peptide recognized with Ld by cytolytic T cells. Cell. 1989;58(2):1–10. doi:10.1016/0092-8674(89)90844-1.
- Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. 2007;117(5):1137–1146. doi:10.1172/JCI31405.
- Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205–214. doi:10.1016/j.cell.2015.03.030.
- Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. 2018;8:86. doi:10.3389/fonc.2018.00086.
- Aldous AR, Dong JZ. Personalized neoantigen vaccines: A new approach to cancer immunotherapy. Bioorg Med Chem. 2018;26(10):2842–2849. doi:10.1016/j.bmc.2017.10.021.
- Heuts J, Varypataki EM, van der Maaden K, Romeijn S, Drijfhout JW, van Scheltinga AT, Ossendorp F, Jiskoot W. Cationic liposomes: A flexible vaccine delivery system for physicochemically diverse antigenic peptides. Pharm Res. 2018;35(11):207. doi:10.1007/s11095-018-2490-6.
- Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515(7528):572–576. doi:10.1038/nature14001.
- Bekri S, Uduman M, Gruenstein D, Mei AHC, Tung K, Rodney-Sandy R, Bogen B, Buell J, Stein R, Doherty K, et al. Neoantigen synthetic peptide vaccine for multiple myeloma elicits T cell immunity in a pre-clinical model. Blood. 2017;130(Suppl 1):1868.
- Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520(7549):692–696. doi:10.1038/nature14426.
- Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534(7607):396–401. doi:10.1038/nature18300.
- Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–226. doi:10.1038/nature23003.
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–221. doi:10.1038/nature22991.
- Tang DC, DeVit M, Johnston SA. Genetic immunization is a simple method for eliciting an immune response. Nature. 1992;356(6365):152–154. doi:10.1038/356152a0.
- Ulmer JB, Donnelly JJ, Parker SE, Rhodes GH, Felgner PL, Dwarki VJ, Gromkowski SH, Deck RR, DeWitt CM, Friedman A. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science. 1993;259(5102):1745–1749. doi:10.1126/science.8456302.
- Wang B, Ugen KE, Srikantan V, Agadjanyan MG, Dang K, Refaeli Y, Sato AI, Boyer J, Williams WV, Weiner DB. Gene inoculation generates immune responses against human immunodeficiency virus type 1. Proc Natl Acad Sci U S A. 1993;90(9):4156–4160. doi:10.1073/pnas.90.9.4156.
- Fioretti D, Iurescia S, Fazio VM, Rinaldi M. DNA vaccines: developing new strategies against cancer. J Biomed Biotechnol. 2010;2010:174378. doi:10.1155/2010/174378.
- Raz E, Carson DA, Parker SE, Parr TB, Abai AM, Aichinger G, Gromkowski SH, Singh M, Lew D, Yankauckas MA. Intradermal gene immunization: the possible role of DNA uptake in the induction of cellular immunity to viruses. Proc Natl Acad Sci U S A. 1994;91(20):9519–9523. doi:10.1073/pnas.91.20.9519.
- Williams BB, Wall M, Miao RY, Williams B, Bertoncello I, Kershaw MH, Mantamadiotis T, Haber M, Norris MD, Gautam A, et al. Induction of T cell-mediated immunity using a c-Myb DNA vaccine in a mouse model of colon cancer. Cancer Immunol Immunother. 2008;57(11):1635–1645. doi:10.1007/s00262-008-0497-2.
- Porgador A, Irvine KR, Iwasaki A, Barber BH, Restifo NP, Germain RN. Predominant role for directly transfected dendritic cells in antigen presentation to CD8+ T cells after gene gun immunization. J Exp Med. 1998;188(6):1075–1082. doi:10.1084/jem.188.6.1075.
- Luxembourg A, Hannaman D, Ellefsen B, Nakamura G, Bernard R. Enhancement of immune responses to an HBV DNA vaccine by electroporation. Vaccine. 2006;24(21):4490–4493. doi:10.1016/j.vaccine.2005.08.014.
- Bins AD, Jorritsma A, Wolkers MC, Hung C-F, Wu T-C, Schumacher TNM, Haanen JBAG. A rapid and potent DNA vaccination strategy defined by in vivo monitoring of antigen expression. Nat Med. 2005;11(8):899–904. doi:10.1038/nm1264.
- Duperret EK, Perales-Puchalt A, Stoltz R, G H H, Mandloi N, Barlow J, Chaudhuri A, Sardesai NY, Weiner DB. A synthetic DNA, multi-neoantigen vaccine drives predominately MHC class I CD8+ T-cell responses, impacting tumor challenge. Cancer Immunol Res. 2019;7(2):174–182. doi:10.1158/2326-6066.CIR-18-0283.
- Falo LD Jr., Kovacsovics-Bankowski M, Thompson K, Rock KL. Targeting antigen into the phagocytic pathway in vivo induces protective tumour immunity. Nat Med. 1995;1:649–653.
- Winzler C, Rovere P, Rescigno M, Granucci F, Penna G, Adorini L, Zimmermann VS, Davoust J, Ricciardi-Castagnoli P. Maturation stages of mouse dendritic cells in growth factor-dependent long-term cultures. J Exp Med. 1997;185(2):317–328. doi:10.1084/jem.185.2.317.
- Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity. 1997;6:715–726.
- Endmann A, Baden M, Weisermann E, Kapp K, Schroff M, Kleuss C, Wittig B, Juhls C. Immune response induced by a linear DNA vector: influence of dose, formulation and route of injection. Vaccine. 2010;28(21):3642–3649. doi:10.1016/j.vaccine.2010.03.034.
- Fransen MF, Schoonderwoerd M, Knopf P, Camps MG, Hawinkels LJ, Kneilling M,van Hall T, Ossendorp F. Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI Insight. 2018;3:23. doi:10.1172/jci.insight.97941.
- McCluskie MJ, Brazolot Millan CL, Gramzinski RA, Robinson HL, Santoro JC, Fuller JT, Widera G, Haynes JR, Purcell RH, Davis HL. Route and method of delivery of DNA vaccine influence immune responses in mice and non-human primates. Mol Med. 1999;5:287–300.
- Kleinovink JW, Marijt KA, Schoonderwoerd MJA, van Hall T, Ossendorp F, Fransen MF. PD-L1 expression on malignant cells is no prerequisite for checkpoint therapy. Oncoimmunology. 2017;6(4):e1294299. doi:10.1080/2162402X.2017.1294299.
- Sato Y, Roman M, Tighe H, Lee D, Corr M, Nguyen MD, Silverman GJ, Lotz M, Carson DA, Raz E. Immunostimulatory DNA sequences necessary for effective intradermal gene immunization. Science. 1996;273(5273):352–354. doi:10.1126/science.273.5273.352.
- Klinman DM, Yamshchikov G, Ishigatsubo Y. Contribution of CpG motifs to the immunogenicity of DNA vaccines. J Immunol. 1997;158:3635–3639.
- Suschak JJ, Wang S, Fitzgerald KA, Lu S. A cGAS-independent STING/IRF7 pathway mediates the immunogenicity of DNA vaccines. J Immunol. 2016;196(1):310–316. doi:10.4049/jimmunol.1501836.
- Jalah R, Patel V, Kulkarni V, Rosati M, Alicea C, Ganneru B, von Gegerfelt A, Huang W, Guan Y, Broderick KE, et al. IL-12 DNA as molecular vaccine adjuvant increases the cytotoxic T cell responses and breadth of humoral immune responses in SIV DNA vaccinated macaques. Hum Vaccin Immunother. 2012;8(11):1620–1629. doi:10.4161/hv.21407.
- Leachman SA, Tigelaar RE, Shlyankevich M, Slade MD, Irwin M, Chang E, Wu TC, Xiao W, Pazhani S, Zelterman D, et al. Granulocyte-macrophage colony-stimulating factor priming plus papillomavirus E6 DNA vaccination: effects on papilloma formation and regression in the cottontail rabbit papillomavirus–rabbit model. J Virol. 2000;74(18):8700–8708. doi:10.1128/jvi.74.18.8700-8708.2000.
- Qing Y, Chen M, Zhao J, Hu H, Xu H, Ling N, Peng M, Ren H. Construction of an HBV DNA vaccine by fusion of the GM-CSF gene to the HBV-S gene and examination of its immune effects in normal and HBV-transgenic mice. Vaccine. 2010;28(26):4301–4307. doi:10.1016/j.vaccine.2010.04.023.
- Lindencrona JA, Preiss S, Kammertoens T, Schüler T, Piechocki M, Wei W-Z, Seliger B, Blankenstein T, Kiessling R. CD4+ T cell-mediated HER-2/neu-specific tumor rejection in the absence of B cells. Int J Cancer. 2004;109(2):259–264. doi:10.1002/ijc.11654.
- Kim JJ, Bagarazzi ML, Trivedi N, Hu Y, Kazahaya K, Wilson DM, Ciccarelli R, Chattergoon MA, Dang K, Mahalingam S, et al. Engineering of in vivo immune responses to DNA immunization via codelivery of costimulatory molecule genes. Nat Biotechnol. 1997;15(7):641–646. doi:10.1038/nbt0797-641.
- Agadjanyan MG, Kim JJ, Trivedi N, Wilson DM, Monzavi-Karbassi B, Morrison LD, Nottingham LK, Dentchev T, Tsai A, Dang K, et al. CD86 (B7-2) can function to drive MHC-restricted antigen-specific CTL responses in vivo. J Immunol. 1999;162:3417–3427.
- Mendoza RB, Cantwell MJ, Kipps TJ. Immunostimulatory effects of a plasmid expressing CD40 ligand (CD154) on gene immunization. J Immunol. 1997;159:5777–5781.
- Ossendorp F, Mengede E, Camps M, Filius R, Melief CJ. Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors. J Exp Med. 1998;187(5):693–702. doi:10.1084/jem.187.5.693.
- Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18(10):635–647. doi:10.1038/s41577-018-0044-0.
- Perez-Diez A, Joncker NT, Choi K, Chan WFN, Anderson CC, Lantz O, Matzinger P. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood. 2007;109(12):5346–5354. doi:10.1182/blood-2006-10-051318.
- Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon E-KM, Muranski P, Restifo NP, Antony PA. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med. 2010;207(3):651–667. doi:10.1084/jem.20091921.
- Elnekave M, Furmanov K, Nudel I, Arizon M, Clausen BE, Hovav AH. Directly transfected langerin+ dermal dendritic cells potentiate CD8+ T cell responses following intradermal plasmid DNA immunization. J Immunol. 2010;185(6):3463–3471. doi:10.4049/jimmunol.1001825.
- Bedoui S, Davey GM, Lew AM, Heath WR. Equivalent stimulation of naive and memory CD8 T cells by DNA vaccination: a dendritic cell-dependent process. Immunol Cell Biol. 2009;87(3):255–259. doi:10.1038/icb.2008.105.
- Gurunathan S, Klinman DM, Seder RA. DNA vaccines: immunology, application, and optimization*. Annu Rev Immunol. 2000;18:927–974. doi:10.1146/annurev.immunol.18.1.927.
- Xiong H, Mittman S, Rodriguez R, Pacheco-Sanchez P, Moskalenko M, Yang Y, Elstrott J, Ritter AT, Müller S, Nickles D, et al. Coexpression of inhibitory receptors enriches for activated and functional CD8+ T cells in murine syngeneic tumor models. Cancer Immunol Res. 2019;7(6):963–976. doi:10.1158/2326-6066.CIR-18-0750.
- Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, Selmi A, Diken M, Boegel S, Paret C, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72(5):1081–1091. doi:10.1158/0008-5472.CAN-11-3722.
- Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie W-R, Hildebrand WH, Mardis ER, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6236):803–808. doi:10.1126/science.aaa3828.
- Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, Zhang W, Sougnez C, Cibulskis K, Sidney J, et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood. 2014;124(3):453–462. doi:10.1182/blood-2014-04-567933.
- Vormehr M, Reinhard K, Blatnik R, Josef K, Beck JD, Salomon N, Suchan M, Selmi A, Vascotto F, Zerweck J, et al. A non-functional neoepitope specific CD8 + T-cell response induced by tumor derived antigen exposure in vivo. Oncoimmunology. 2019;8(3):1553478. doi:10.1080/2162402X.2018.1553478.