
ABSTRACT
Systemic treatment with the active transcription inhibitor lurbinectedin aims at inducing tumor cell death in hyperproliferative neoplasms. Here we show that cell death induced by lurbinectedin reinstates and enhances systemic anticancer immune responses. Lurbinectedin treatment showed traits of immunogenic cell death, including the exposure of calreticulin, the release of ATP, the exodus of high mobility group box 1 (HMGB1) and type 1 interferon responses in vitro. Lurbinectedin treated cells induced antitumor immunity when injected into immunocompetent animals and treatment of transplanted fibrosarcomas reduced tumor growth in immunocompetent yet not in immunodeficient hosts. Anticancer effects resulting from lurbinectedin treatment were boosted in combination with PD-1 and CTLA-4 double immune checkpoint blockade (ICB), and lurbinectedin combined with double ICB exhibited strong antineoplastic effects. Cured animals exhibited long term immune memory effects that rendered them resistant to rechallenge with syngeneic tumors underlining the potency of combination therapy with lurbinectedin.
Introduction
Primary or transplantable tumors react to anthracycline-based chemotherapy with durable response in syngeneic immunocompetent mice yet fail to do so in immunodeficient hosts.Citation1–Citation3 Consistently, retrospective clinical studies in patients with solid tumors subjected to chemotherapy showed that severe lymphopenia negatively affects prognosis,Citation4,Citation5 which points to the fact that chemotherapy-elicited anticancer immunity plays a critical role for the outcome of anticancer therapy.Citation6,Citation7 Based on these findings,Citation1–Citation3 we introduced the hypothesis that some chemotherapeutic agents can induce immunogenic cell death (ICD) in tumors and convert them into a therapeutic vaccine, hence stimulating an immune response that can control residual cancer cells.
Selected chemotherapeutics such as anthracyclines and oxaliplatin are able to induce ICDCitation1–Citation3 while many other antineoplastic agents including cisplatin and mitomycin C fail to do so. Cancer cells undergoing ICD can evoke anticancer immunity and protect against a subsequent challenge with living cells exhibiting the same antigenic profile in miceCitation1–Citation3 or elicit anticancer immune responses during chemotherapy in patients.Citation8 Distinctive properties of immunogenic cell death include the exposure of calreticulin (CALR) at the cytoplasmic surface,Citation3,Citation8,Citation9 the autophagy-dependent liberation of ATP from stressed and dying cells,Citation10,Citation11 the cell death-associated exodus of nuclear high mobility group box 1 (HMGB1)Citation12,Citation13 and the stimulation of an autocrine or paracrine type-1 interferon response.Citation14 CALR serves as a de novo uptake signal and stimulates the engulfment of dying cancer cells by dendritic cells (DCs).Citation3 HMGB1 binds to toll-like receptor-4 (TLR4) entities on DC, eliciting MYD88-dependent signaling that facilitates tumor antigen processing.Citation3,Citation15 ATP ligates purinergic receptors of the P2X type and thus activates the NLRP3 inflammasome to stimulate the production of interleukin-1β (IL-1β) by DC and eventually interferon-γ (IFNγ) by CD8+ cytotoxic T lymphocytes (CTL).Citation10,Citation16
The sum of danger associated molecular patterns (DAMP) emitted during ICD is necessary to generate anticancer immunity, thus tumors growing in Tlr4−/-, P2rx7−/-, Myd88−/-, Nlrp3−/-, Il1r−/-, Ifnγ−/-, Ifnγr−/-, Fpr1−/-, athymic or CD8+ T cell-depleted mice fail to respond to immunogenic chemotherapeutic regimens. Loss-of-function mutations of FPR1, P2RX7 or TLR4 in breast cancer are negatively correlated with clinical response to adjuvant chemotherapy with anthracyclines.Citation3,Citation10,Citation13,Citation14,Citation17–Citation19 These results imply the obligate contribution of anticancer immune responses to the success of ICD-inducing chemotherapies.
Lurbinectedin is a selective inhibitor of active transcription of protein-coding genesCitation20 that is currently undergoing clinical investigation and has recently gained orphan drug approval for the treatment of small cell lung cancer (SCLC). Here, we investigated the capacity of lurbinectedin to stimulate the emission of immunogenic DAMPs and tested anticancer immune responses in three experimental in vivo models. Our results support the contention that lurbinectedin causes immunogenic cell death in tumors and creates anticancer immunity.
Results and discussion
Emission of immunogenic signals by lurbinectedin
The known parameters determining ICD are the translocation of CALR to the surface of the plasma membrane, the autophagy-dependent liberation of ATP and the release of the non-histone binding protein HMGB1, which occur before, during and after apoptosis, respectively. The production of type I interferons (IFNs) has been added to the list of ICD hallmarks as it controls autocrine or paracrine circuits that underlie cancer immunosurveillance.
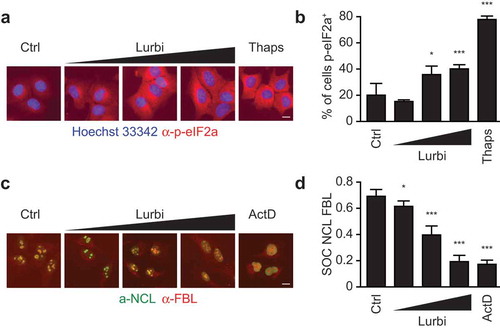
In a systematic screening campaign, the capacity of lurbinectedin to induce immunogenic cell death in cancer cells was assessed in human osteosarcoma U2OS cells stably expressing fluorescent biosensors for the detection of CALR-relocation (as a surrogate marker for CALR surface exposure), HMGB1 release and Type I IFN responses together with U2OS WT cells stained with the ATP-sensitive dye quinacrine. ICD-related parameters were measured at 4, 8, 16 and 32 hours post exposure to lurbinectedin from 1 nM to 1 µM by robotized epifluorescence microscopy followed by automated image analysis (). The induction of cell death was evaluated based on changes in the nuclear morphology visualized by means of the DNA intercalating dye Hoechst 33342. Lurbinectedin caused a dose- and time-dependent drop in cellular viability comparable to mitoxantrone (MTX) that was used at 1 and 3 µM as a positive control throughout all experiments. The translocation of a CALR-GFP (green fluorescent protein) fusion protein from the perinuclear ER to the cellular periphery was measured by assessing cytoplasmic “granularity” (see Materials and Methods) as an indicator for the formation of CALR-containing vesicles and as a surrogate marker for CALR exposure. Lurbinectedin, similar to MTX, induced a time- and dose-dependent increase in CALR-granularity as compared to untreated controls. The reduction of intracellular ATP (as an indicator for ATP release) was assessed by measuring the decrease in the cytoplasmic granularity of ATP containing vesicles stained with the fluorescent probe quinacrine. As compared to untreated controls a significant decrease in ATP signal similar to MTX was detectable for lurbinectedin. The effect was dose-dependent and decreased over time in line with the fragile nature of the metabolite. HMGB1 release was detected as a loss in the nuclear fluorescence of an HMGB1-GFP chimera. A significant decrease in nuclear GFP signal was detected for MTX and lurbinectedin at medium to late time points. Type I interferon (IFN) production was measured using U2OS biosensor cells stably expressing a GFP under the control of the MX1 (a Type I IFN response gene) promoter. To this aim the supernatant of U2OS cells following treatment and additional 48 hours incubation with fresh media was used to treat the biosensor cells. Following the type 1 IFN response was monitored by means an increase in GFP fluorescence intensity. A significant increase in de novo GFP signal intensity was detected for both lurbinectedin and MTX throughout all time points ()). Similar results were obtained when the approach was repeated in human breast cancer HCC70 cells ()), human colon carcinoma HT29 ()) and mouse fibrosarcoma MCA205 cells ()). Next, we investigated the capacity of lurbinectedin to activate two additional characteristics of common ICD inducers, the phosphorylation of the eukaryotic translation initiation factor 2 alpha (eIF2α) and the inhibition of general transcription. Indeed, lurbinectedin led to a dose-dependent phosphorylation of eIF2α monitored by fluorescence microscopy upon immunostaining with a phosphoneoepitope-specific antibody (,)). Lurbinectedin also inhibited mRNA transcription at a level comparable to a known transcription-inhibitor, as assessed by visualizing the dissociation of nucleolin and fibrillarin by microscopy (,)), an accepted proxy of suppressed transcription.Citation21 Lurbinectedin holds many of the described in vitro parameters of ICD, thus qualifying for further in vivo investigations in immunocompetent animals, which remains the gold standard assay for the determination of ICD-mediated anticancer immunity.
Figure 1. Immunogenic cell death assessment in solid tumors.
(a) Human osteosarcoma U2OS (a), human breast cancer HCC70 (b) human colon cancer HT29 cells (c) and murine methylcholantrene-induced fibrosarcoma MCA205 cells (d) were treated with lurbinectedin (Lurbi, 1 nM, 10 nM, 100 nM and 1 µM) for the indicated times. Subsequently, the cells were stained with 1 µM Hoechst 33342 and 1 µM propidium iodide and assessed for the loss of viability by automated image acquisition. Images were segmented, cellular debris was excluded and the number of cells with normal nuclear morphology was enumerated. Cells stably expressing CALR-GFP were treated as above. Following the cells were fixed with 3.7% of PFA, stained with 1 µM Hoechst 33342 and assessed by automated image acquisition. Images were segmented, cellular debris was excluded and CALR-GFP granularity (a surrogate marker of CALR exposure) was evaluated in the cytoplasmic region of cells with normal nuclear morphology. Wild type cells were treated as above and then assessed for cytoplasmic quinacrine granularity (after staining with the ATP-sensitive dye quinacrine together with Hoechst 33342) by automated image acquisition, segmentation and analysis. Cells stably expressing HMGB1-GFP were treated as above and then assessed for nuclear HMGB1-GFP fluorescence intensity. The cells were fixed and stained with Hoechst 33342 and images were acquired, segmented and analyzed. WT cells were treated as above and following the media was changed and the cells were incubated for 48 hours before the supernatant was used to treat MX1-GFP biosensor cells for additional 48 hours. The cells were fixed and stained with Hoechst 33342 before type 1 IFN responses were monitored by means of automated microscopy as an increase in GFP fluorescence intensity. Mitoxantrone (MTX, 1 and 3 µM) was used as a positive control. The means of quadruplicate assessments and p-values are depicted as heat maps. (*p < .01; **p < .005; ***p < .001, two-tailed Student’s t test).
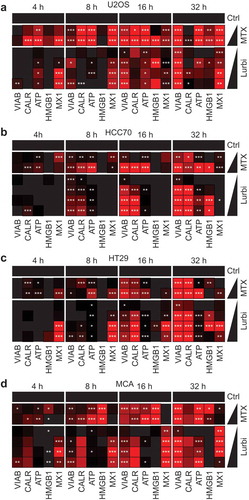
Figure 2. Traits of immunogenic cell death.
Human osteosarcoma U2OS cells were treated with 10, 50 or 100 nM lurbinectedin (Lurbi) for 6 hours. Thapsigargin (Thaps, 3 µM) was used as a positive control. The cells were fixed with 3.7% PFA and DNA was stained with 1 µM Hoechst 33342. Following the phosphorylation of the eukaryotic translation initiation factor 2 alpha (eIF2a) was assessed with phosphoneoepitope-specific antibody and was monitored by means of automated microscopy as an increase in cytoplasmic fluorescence intensity. (a,b) The level of transcription was measured in U2OS cell treated as above with Lurbi. The transcription inhibitor actinomycin D (ActD) was used as a control. The cells were fixed as above and following the colocalization of nucleolin and fibrillarin was assessed as an indicator for transcriptional activity (c,d) Scale bar equals 10 µm and bar charts depict depict mean values ± SD of quadruplicate assessments (*p < .01; ***p < .001, two-tailed Student’s t test).
Anticancer immunity induced by lurbinectedin
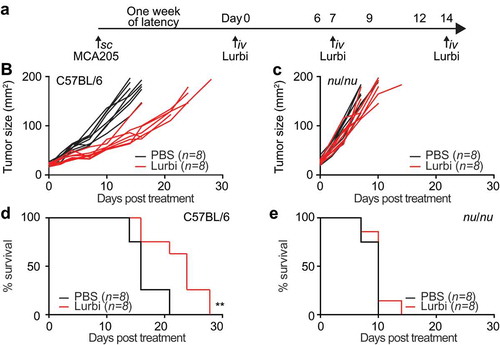
In order to assess the capacity of lurbinectedin to stimulate anticancer immunity in a monotherapeutic approach and to convert tumor cells into a therapeutic vaccine we exposed murine fibrosarcoma cells to the drug in vitro (in conditions previously established to induce a sufficient amplitude of cell death) and then injected the dying cancer cells into syngeneic immunocompetent mice. One week later, the animals were re-challenged injecting live tumor cells of the same kind into the opposite flank, ()). In this setting, a decrease of tumor growth can be interpreted as sign of a productive anticancer immune response. Indeed, lurbinectedin-treated cells significantly reduced tumor growth (p = .0094) ()) and led to an increase in overall survival ()). As compared to know ICD inducersCitation1–Citation3 the vaccination effects observed here were rather limited yet statistically significant. Next we evaluated the effect of lurbinectedin on established cancers growing on immunocompetent or immunodeficient mice. MCA 205 tumors were implanted subcutaneously on immunocompetent C57BL/6 as well as in athymic nu/nu mice. When the tumors became palpable, the animals were treated with three consecutive intravenous injections of 0.18 mg/kg lurbinectedin on day 1, 7 and 14. ()). The treatment with lurbinectedin had significant therapeutic benefit in immunocompetent animals. The tumor growth was significantly reduced as compared to control animals (p < .0001) ()) and overall survival was increased ()). This effect was exclusively observed when tumors grew on immunocompetent mice, yet was lost when the tumors proliferated on athymic (nu/nu) mice (,)). These results underscore the obligate contribution of the immune system to the chemotherapeutic activity of lurbinectedin.
Figure 3. Anticancer vaccination efficacy of lurbinectedin-treated cells.
MCA205 cells treated for 20 h with 1 µM lurbinectedin were inoculated subcutaneously (s.c.) into immunocompetent C57BL/6 mice, which were rechallenged 7 days later s.c. with living cells of the same type. The tumor growth was measured until endpoints were reached and overall survival was evaluated regularly for the following 30 days (n = 6). (*p < .01, two-tailed Student’s t test, compared to all other groups). Data were analyzed with TumGrowth.
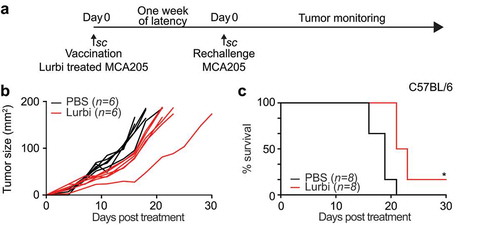
Figure 4. Therapeutic efficacy of lurbinectedin in immunocompetent and immunodeficient hosts.
Live MCA205 cells were injected subcutaneously (s.c.) into immunocompetent C57BL/6 mice or immunodeficient nu/nu mice as depicted in the scheme in (a) When tumors became palpable, mice were intravenously (i.v.) injected with 0.14 mg/Kg lurbinectedin (on day 1,7 and 14). Tumor growth was assessed regularly for the following 30 days. Data is depicted as tumor growth curves (b,d) and overall survival plots (c,e). Data were analyzed with TumGrowth.
Combinatorial effects of lurbinectedin and αPD-1/αCTLA-4 double immune checkpoint blockade
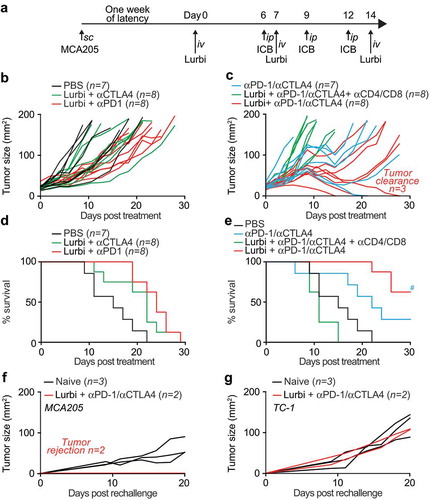
Given the capacity of lurbinectedin to induce immune-dependent anticancer effects on established tumors, we investigated whether this agent could sensitize cancers to therapy with immune checkpoint blockers targeting CTLA-4 or PD-1. For this, established MCA205 fibrosarcomas were treated with Lurbinectedin as before and subjected to immunotherapy with antibodies specific for CTLA-4, PD-1 or a combination of both on day 6, 9 and 12, when the anticancer immune response in the tumor peaks ()). Tumor monitoring led to the deduction that the most efficient therapeutic regimen was a combination of all three anticancer agents (lurbinectedin, αCTLA-4 and αPD-1) in contrast to single-ICB therapies that appeared to be relatively inefficient in this setting (–)). The combination of lurbinectedin with αCTLA-4/αPD-1 dual checkpoint blockade in tumor-bearing animals significantly extended life expectancy and, moreover, led to tumor clearance in 3/8 mice in the time frame of the experiment ()). The effect of lurbinectedin with αCTLA-4/αPD-1 dual checkpoint blockade was abrogated in conditions in which CD4+ and CD8+ cytotoxic T lymphocytes (CTLs) were depleted. Mice that had been rendered tumor-free for more than 50 days rejected tumors upon rechallenge with the same cancer cell type from which they had been cured (MCA205), yet developed cancers when rechallenged with TC1 tumor cells (,)). Thus, mice that had been cured by a combination of systemic lurbinectedin-based chemotherapy and immunotherapy had established a specific anticancer immune response that generated immunological memory.
Figure 5. Sequential lurbinectedin treatment with double immune checkpoint blockade exhibits systemic antitumor immunityC57BL/6 mice were inoculated subcutaneously (s.c.) with murine fibrosarcoma MCA205. Palpable tumors were treated with sequential intravenous (i.v.) injections of 0.14 mg/Kg lurbinectedin (Lurbi) as indicated in (a). Single- or double-immune checkpoint blockade was mounted by sequential intraperitoneal (i.p.) injections of monoclonal antibodies targeting CTLA-4 or PD-1 at day 6, 9 and 12 post treatment and tumor growth (b,c) and overall survival (d,e) were assessed regularly for the following 30 days. (f,g) The generation of immunological memory was assessed in cured animals by rechallenged with MCA205 and TC-1. Naïve animals were used as controls. Individual tumor growth curves are depicted. Data were analyzed with TumGrowth.
Lurbinectedin retards the growth of carcinogen-induced and spontaneous breast cancer
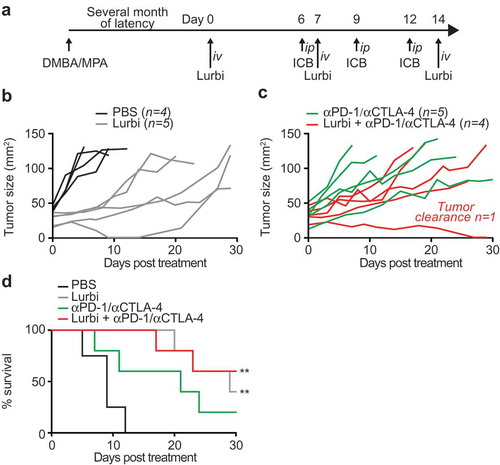
To explore the potential lurbinectedin for the therapy of breast cancer, we took advantage of a hormone/carcinogen induced breast cancer model activated by the continuous stimulation of progesterone receptors by medroxyprogesterone acetate (MPA) and the repeated exposure to the DNA-damaging agent dimethylbenzantracene (DMBA). This induced model of breast cancer is known to be modulated by the immune system.Citation22 We treated mice with palpable MPA/DMBA-induced tumors by systemic injection with lurbinectedin alone or in combination with double immune checkpoint blockade neutralizing CTLA-4 and PD-1 ()). Both interventions significantly reduced tumor growth and increased overall survival. However, only the combination with αCTLA-4/αPD-1 yielded tumor clearance in the time frame of the experiment (–)).
Figure 6. Lurbinectedin retards the growth of spontaneous tumors.
Medroxyprogesterone acetate (MPA) pellets (50 mg, 90-day release) were implanted subcutaneously into the interscapular area of immunocompetent C57BL/6 mice. Then the animals received 1 mg dimethylbenzantracene (DMBA) administered by oral gavage 6 x during 7 weeks. When spontaneous tumors became palpable mice were randomly assigned to receive 0,14 mg/Kg lurbinectedin (Lurbi) alone or in combination with double immune checkpoint blockade with monoclonal antibodies targeting CTLA-4 and PD-1 at day 6, 9 and 12 post treatment (a). The tumor area and overall survival were measured regularly until ethical endpoints were reached (b,c,d). Data were analyzed with TumGrowth (https://github.com/kroemerlab).
Concluding remarks
The results of this study suggest that lurbinectedin efficiently induces cell death in a broad panel of solid tumors. This procedure likely does not only cause the cells to succumb to disintegration but rather triggers traits of immunogenic cell death, including the phosphorylation of eIF2a and the release of danger associated molecular patterns (DAMPs). Irrespective of the exact molecular mechanisms accounting for these effects, there are a number of evidences advocating for lurbinectedin-triggered cancer-specific immunogenicity. Thus, animals that had been cured by lurbinectedin from established cancers became resistance to rechallenge with the same cancer type. The therapeutic effect of lurbinectedin was neutralized in conditions in which either the host was immunocompromised or T-cell had been depleted. Furthermore, the recapitulation in a heterogeneous spontaneous tumor model of effects that were previously observed in homogenous transplanted tumors indicates that the results presented here hold a high translational value.
Altogether, these results convincingly demonstrate that lurbinectedin mediated immunochemotherapy may be advantageously combined with clinically established immune checkpoint blockade regimens.
Materials & methods
Cell culture and chemicals
All media and cell culture supplements were from Thermo Fisher Scientific (Carlsbad, CA, US). Lurbinectedin was provided by PharmaMar (Madrid, Spain). Cell culture plastics and consumables were purchased from Greiner Bio-One (Kremsmünster, Austria). Human osteosarcoma U2OS cells previously genetically altered as described earlier,Citation23 murine methylcholanthrene-induced fibrosarcoma MCA-205 cells and murine lung cancer TC-1 cells were cultured in Glutamax®-containing DMEM medium supplemented with 10% fetal bovine serum (FBS), and 10 mM HEPES. Cells were cultured in a temperature-controlled environment at 37°C with a humidified atmosphere containing 5% CO2.
Automated image acquisition and analysis
One day before the experiment 5 × 103 cells were seeded in 96-well µClear imaging plates (Greiner BioOne) and let adhere under standard culture conditions. The following day cells were treated with lurbinectedin at 0.001, 0.01, 0.1 and 1 µM for 4, 8, 16 or 32 hours. Then cells were fixed with 3.7% formaldehyde supplemented with 1 μg/ml Hoechst 33342 for 30 min at RT. The fixative was changed to PBS and the plates were analyzed by automated microscopy. For the detection of ATP enriched vesicles, the cells were labeled after 4, 8, 16 or 32 hours of incubation with the fluorescent dye quinacrine (as described beforeCitation23). In short, cells were incubated with 5 µm quinacrine and 1 µg/ml Hoechst 33342 in Krebs-Ringer solution (125 mM NaCl, 5 mM KCl, 1 mM MgSO4, 0.7 mM KH2PO4, 2 mm CaCl2, 6 mM glucose and 25 mM Hepes, pH 7.4) for 30 minutes at 37°C. Thereafter, cells were rinsed with Krebs-Ringer and viable cells were microscopically examined. For automated fluorescence microscopy a robot-assisted Molecular Devices IXM XL BioImager (Molecular Devices, Sunnyvale, CA, USA) equipped with SpectraX light source (Lumencor, Beaverton, OR, USA), adequate excitation and emission filters (Semrock, Rochester, NY, USA) and a 16-bit monochromes sCMOS PCO.edge 5.5 camera (PCO Kelheim, Germany) and a 20 X PlanAPO objective (Nikon, Tokyo, Japan) was used to acquire a minimum of 9 view fields, followed by automated image processing with the custom module editor within the MetaXpress software (Molecular Devices). Depending on the utilized biosensor cell line the primary region of interest (ROI) was defined by a polygon mask around the nucleus allowing for the enumeration of cells, the detection of morphological alterations of the nucleus and nuclear fluorescence intensity. Cellular debris was excluded from the analysis and secondary cytoplasmic ROIs were used for the quantification of CALR-GFP or quinacrine containing vesicles. For the latter, the images were segmented and analyzed for GFP granularity by comparing the standard deviation of the mean fluorescence intensity of groups of adjacent pixels (coefficient of variation) within the cytoplasm of each cell to the mean fluorescence intensity in the same ROI using the MetaXpress software (Molecular Devices).
In vivo experimentation
Six- to eight-week-old female wild-type C57BL/6 and nu/nu mice were obtained from Envigo France (Huntingdon, UK) and were kept in the animal facility at the Gustave Roussy Campus Cancer in a specific pathogen–free and temperature-controlled environment with 12 h day, 12 h night cycles and received food and water ad libitum. Animal experiments were conducted in compliance with the EU Directive 63/2010 and protocols 2013_094A and were approved by the Ethical Committee of the Gustave Roussy Campus Cancer (CEEA IRCIV/IGR no. 26, registered at the French Ministry of Research). As described,Citation24,Citation25 MCA205 tumors were established in C57BL/6 mice by subcutaneously (s.c.) injection of 5 × 105 cells. When tumors became palpable, 0.18 mg/Kg lurbinectedin was injected sequentially once a week intravenously into the tail vein and animal well-being and tumor growth were monitored. A total of 0.5 mg of anti-CD8 (clone 2.43 BioXCell BE0061) and anti-CD4 (clone GK1.5 BioXCell BE0003-1) intraperitoneal (i.p.) injections were repeated every 7 days to assure the complete depletion of both T cell populations during the whole experiment. Mice were sacrificed when tumor size reached end-point or signs of obvious discomfort associated to the treatment were observed following the EU Directive 63/2010 and our Ethical Committee advice. Tumor-free animals were kept for more than 30 days before testing the generation of immunological memory by s.c. rechallenge with 5 × 105 TC-1 in one flank and 5 × 105 MCA205 cells injected in the contralateral flank. Animals were monitored and tumor growth documented regularly until end-points were reached. Statistical analysis was performed by applying 2-way ANOVA analysis followed by Bonferroni’s test comparing to control conditions (* p < .05, ** p < .01 and ***p < .001). Murine fibrosarcoma MCA205 cells were incubated with 1 µM lurbinectedin for 24 h, resulting in approximately 70% cell death. For vaccination experiments, 3 × 105 dying MCA205 cells were inoculated s.c. into the left flanks of six-week-old female C57BL/6 mice. Seven to ten days later, animals were re-challenged in the opposite flank with 3 × 105 living MCA205 cells, and tumor growth and incidence were monitored. Six-week-old female C57BL/6 mice (n = 12 per group) underwent surgical implantation of slow-release medroxyprogesterone acetate (MPA) pellets (50 mg, 90-day release; Innovative Research of America, Sarasota, Fl, US) s.c. Two-hundred μL of 5 mg/mL dimethylbenzantracene (DMBA, Sigma Aldrich, St. Louis, MO, US) dissolved in corn oil was administered by oral gavage once per week for 7 weeks.
Immune checkpoint blockade
Double or single immune checkpoint blockade was applied by repeated intraperitoneal injections of monoclonal antibody specific to PD-1 (200 µg, Clone 29F.1A12, BioXcell, West Lebanon, NH, USA) or CTLA-4 (200 µg, Clone 9D9, BioXcell) at day 6, 9 and 12 upon initiation of the treatment with lurbinectedin. Animals were monitored regularly and the tumor growth was documented until ethical end-points were reached. Statistical analysis was performed employing 2-way ANOVA analysis followed by Bonferroni’s test comparing to control conditions (* p < .05, ** p < .01 and ***p < .001).
Statistical procedures
Unless otherwise specified, experiments were performed in quadruplicate instances. Data were analyzed with the freely available software R (https://www.r-project.org). Significances were calculated using a student t-test with Welch correction. Thresholds for each assay were applied based on the Gaussian distribution of positive and negative controls. In vivo tumor growth was analyzed with the help of the TumGrowth software packageCitation26 freely available at https://github.com/kroemerlab.
Abbreviations
| DAMP | = | danger-associated molecular pattern |
| ICD | = | immunogenic cell death |
Conflict of interest disclosure
This study was supported by Pharmamar. JFM-L is a full-time employee of Pharmamar.
Acknowledgments
WX and YW were supported by the China Scholarship Council, JH is the recipient of a PhD fellowship from the Foundation Philanthropia; GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Gustave Roussy Odyssea, the European Union Horizon 2020 Project Oncobiome; Fondation Carrefour; High-end Foreign Expert Program in China (GDW20171100085 and GDW20181100051), Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and the SIRIC Cancer Research and Personalized Medicine (CARPEM).
References
- Casares N, Pequignot MO, Tesniere A, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202(12):1–9. doi:10.1084/jem.20050915.
- Mattarollo SR, Loi S, Duret H, Ma Y, Zitvogel L, Smyth MJ. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res. 2011;71(14):4809–4820. doi:10.1158/0008-5472.CAN-11-0753.
- Obeid M, Tesniere A, Ghiringhelli F, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54–61. doi:10.1038/nm1523.
- Ray-Coquard I, Cropet C, Van Glabbeke M, et al. Lymphopenia as a prognostic factor for overall survival in advanced carcinomas, sarcomas, and lymphomas. Cancer Res. 2009;69(13):5383–5391. doi:10.1158/0008-5472.CAN-08-3845.
- Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14(12):717–734. doi:10.1038/nrclinonc.2017.101.
- Lesterhuis WJ, Haanen JB, Punt CJ. Cancer immunotherapy–revisited. Nat Rev Drug Discovery. 2011;10(8):591–600. doi:10.1038/nrd3500.
- Zitvogel L, Kepp O, Kroemer G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol. 2011;8(3):151–160. doi:10.1038/nrclinonc.2010.223.
- Zappasodi R, Pupa SM, Ghedini GC, et al. Improved clinical outcome in indolent B-cell lymphoma patients vaccinated with autologous tumor cells experiencing immunogenic death. Cancer Res. 2010;70(22):9062–9072. doi:10.1158/0008-5472.CAN-10-1825.
- Fucikova J, Kralikova P, Fialova A, et al. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011;71(14):4821–4833. doi:10.1158/0008-5472.CAN-11-0950.
- Ghiringhelli F, Apetoh L, Tesniere A, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15(10):1170–1178. doi:10.1038/nm.2028.
- Michaud M, Martins I, Sukkurwala AQ, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334(6062):1573–1577. doi:10.1126/science.1208347.
- Apetoh L, Ghiringhelli F, Tesniere A, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007;220:47–59. doi:10.1111/j.1600-065X.2007.00573.x.
- Tesniere A, Schlemmer F, Boige V, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29(4):482–491. doi:10.1038/onc.2009.356.
- Sistigu A, Yamazaki T, Vacchelli E, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20(11):1301–1309. doi:10.1038/nm.3708.
- Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050–1059. doi:10.1038/nm1622.
- Ma Y, Adjemian S, Mattarollo SR, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38(4):729–741. doi:10.1016/j.immuni.2013.03.003.
- Vacchelli E, Ma Y, Baracco EE, et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science. 2015;350(6263):972–978. doi:10.1126/science.aad0779.
- Kroemer G, Senovilla L, Galluzzi L, Andre F, Zitvogel L. Natural and therapy-induced immunosurveillance in breast cancer. Nat Med. 2015;21(10):1128–1138. doi:10.1038/nm.3944.
- Vacchelli E, Enot DP, Pietrocola F, Zitvogel L, Kroemer G. Impact of pattern recognition receptors on the prognosis of breast cancer patients undergoing adjuvant chemotherapy. Cancer Res. 2016;76(11):3122–3126. doi:10.1158/0008-5472.CAN-16-0294.
- Santamaría Nuñez G, Robles CM, Giraudon C, et al. Lurbinectedin specifically triggers the degradation of phosphorylated RNA polymerase II and the formation of DNA breaks in cancer cells. Mol Cancer Ther. 2016;15(10):2399–2412. doi:10.1158/1535-7163.MCT-16-0172.
- Peltonen K, Colis L, Liu H, et al. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell. 2014;25(1):77–90. doi:10.1016/j.ccr.2013.12.009.
- Pietrocola F, Pol J, Vacchelli E, et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell. 2016;30(1):147–160. doi:10.1016/j.ccell.2016.05.016.
- Martins I, Kepp O, Schlemmer F, et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene. 2011;30(10):1147–1158. doi:10.1038/onc.2010.500.
- Iribarren K, Buque A, Mondragon L, et al. Anticancer effects of anti-CD47 immunotherapy in vivo. Oncoimmunology. 2019;8(3):1550619. doi:10.1080/2162402X.2018.1550619.
- Zhou H, Mondragon L, Xie W, et al. Oncolysis with DTT-205 and DTT-304 generates immunological memory in cured animals. Cell Death Dis. 2018;9(11):1086. doi:10.1038/s41419-018-1127-3.
- Enot DP, Vacchelli E, Jacquelot N, Zitvogel L, TumGrowth: KG. An open-access web tool for the statistical analysis of tumor growth curves. Oncoimmunology. 2018;7(9):e1462431. doi:10.1080/2162402X.2018.1490854.