
ABSTRACT
Chimeric antigen receptor (CAR) T cells have shown impressive therapeutic potential. Due to the lack of direct control mechanisms, therapy-related adverse reactions including cytokine release- and tumor lysis syndrome can even become life-threatening. In case of target antigen expression on non-malignant cells, CAR T cells can also attack healthy tissues. To overcome such side effects, we have established a modular CAR platform termed UniCAR: UniCAR T cells per se are inert as they recognize a peptide epitope (UniCAR epitope) that is not accessible on the surface of living cells. Bifunctional adapter molecules termed target modules (TM) can cross-link UniCAR T cells with target cells. In the absence of TMs, UniCAR T cells automatically turn off. Until now, all UniCAR TMs were constructed by fusion of the UniCAR epitope to an antibody domain. To open up the wide field of low-molecular-weight compounds for retargeting of UniCAR T cells to tumor cells, and to follow in parallel the progress of UniCAR T cell therapy by PET imaging we challenged the idea to convert a PET tracer into a UniCAR-TM. For proof of concept, we selected the clinically used PET tracer PSMA-11, which binds to the prostate-specific membrane antigen overexpressed in prostate carcinoma. Here we show that fusion of the UniCAR epitope to PSMA-11 results in a low-molecular-weight theranostic compound that can be used for both retargeting of UniCAR T cells to tumor cells, and for non-invasive PET imaging and thus represents a member of a novel class of theranostics.
Introduction
Among tumor diseases prostate cancer (PCa) is still one of the major causes of cancer-related death in men worldwide.Citation1,Citation2 Even though primary localized tumors are well manageable, survival benefit of metastatic, castrate-resistant PCa patients is still limited.Citation3–Citation5 In order to improve the clinical outcome of these patients, on the one hand, novel innovative therapy options are needed. On the other hand, accurate imaging tools play a critical role to identify appropriate therapeutic measures.Citation6 Thus, theranostics of PCa based on antibodies (Abs) or small molecules is an uprising and highly promising field.
The prostate-specific membrane antigen (PSMA) represents a valuable molecular target for PCa theranostics. Physiologically, the type II transmembrane glycoprotein PSMA is expressed on healthy prostate and to a lower extent also on other tissues including, e.g. kidneys, intestines, brain, lacrimal and salivary glands.Citation7–Citation9 PSMA levels increase up to 1000-fold on about 90% of primary and metastatic PCa cells including bone and lymph node metastases.Citation7–Citation12 Most importantly, there is a strong correlation of PSMA expression with high tumor-grade and stage as well as metastatic and hormone-refractory disease.Citation13
With the clinical introduction in 2012,Citation14,Citation15 the radiotracer 68Ga-PSMA-11 leveraged PSMA-directed theranostic radiopharmaceuticals.Citation5,Citation16,Citation17 The small peptide ligand PSMA-11 is composed of a Glu-urea-Lys motif as pharmacophore and the 68Ga-complexing agent N,N´-bis[2-hydroxy-5-(carboxyethyl)benzyl]-ethylenediamine-N,N´-diacetic acid (HBED-CC).Citation18 Among other Glu-ureido-based PSMA ligands, 68Ga-PSMA-11 has shown to be a widespread, clinically accepted positron emission tomography (PET) imaging agent especially valuable for detection of lymph node and bone metastases as well as diagnosis of biochemical recurrence with low PSA levels (<2 ng/μL).Citation5,Citation6,Citation17 Further modifications finally resulted in the development of theranostic PSMA radiotracers, e.g. PSMA-617Citation19,Citation20 and PSMA I&T,Citation21 which can be labeled with both diagnostic (e.g. 68Ga) and therapeutic (e.g. 177Lu or 225Ac) radiometals. Depending on the employed radioisotope, these small molecules are successfully utilized in PCa patients for initial diagnosis, re-/staging, and monitoring as well as for salvage radioligand therapy of metastatic, castrate-resistant or progressive disease.Citation6,Citation22
Besides radionuclide therapy, immunotherapeutic approaches based on chimeric antigen receptor (CAR) T cells play an increasingly important role for cancer therapy including PCa.Citation23–Citation35 CARs are genetically engineered synthetic receptors consisting of (i) an extracellular binding moiety most commonly derived from tumor-specific monoclonal Abs, (ii) a transmembrane domain, and (iii) intracellular signaling domain(s) taken from activating and optionally costimulatory immune receptors.Citation23,Citation26,Citation31 After modification of T cells with CARs, they are able to bind tumor-associated antigen (TAA)-expressing cells independently of their MHC restriction, which finally results in T cell-mediated tumor cell killing.Citation23,Citation24,Citation26,Citation31 The pivotal role of CAR T cells for management of tumor diseases is clearly underlined by the approval of two different CD19-specific CAR T cell products in 2017.Citation22,Citation36–Citation38 However, the broad clinical application of these living drugs also taught us that precise control mechanisms of CAR T cells after adoptive transfer will be highly important to improve their safety profile. Major side effects are mainly related to tumor lysis and cytokine release syndrome as well as on-target/off-tumor effects that are associated with the expression of the chosen target structure on healthy tissues.Citation24,Citation38 In order to rule out lifelong, target-specific off-tumor side effects and to facilitate regulation of adoptively transferred CAR T cells much progress is being undertaken to include additional safety switches [e.g. Citation39,Citation40,Citation41,Citation42]. We therefore established a modular platform technology termed UniCAR, which is a two-component system consisting of UniCAR T cells and target modules (TMs).Citation43–Citation50 UniCARs are second-generation CARs, which target the La/SS-B-derived peptide epitope E5B9 (UniCAR epitope).Citation51,Citation52 As this epitope is physiologically not accessible on the surface of intact living cells, adoptively transferred UniCAR T cells are in contrast to TAA-specific CAR T cells per se inactive. Retargeting of UniCAR T cells to tumor cells is facilitated by a separate TM comprising a tumor-specific Ab binding moiety (e.g. single-chain fragment variable (scFv) or nanobody) and the UniCAR epitope (). Thus, UniCAR T cells are engaged for tumor cell killing only in the presence of TMs.Citation43–Citation50 Based on rapid TM elimination, adverse effects of redirected UniCAR T cells should be easily controlled by stopping TM infusion. After recovery of the patient, TM supply and hence immunotherapy can be restarted. The UniCAR platform technology allows moreover multi-tumor targeting approaches simply by combing UniCAR T cells subsequently or simultaneously with two or more TMs. Since the first presentation of the UniCAR idea in 2014,Citation43 several, so far only Ab-based TMs were described including TMs directed against the TAAs CD33, CD123, prostate stem cell antigen, PSMA, epidermal growth factor receptor, GD2, CD19, or STn.Citation43–Citation54
Figure 1. Retargeting of UniCAR T cells with the novel PET tracer-based PSMA PLT-TM. (a) In previous studies, we showed that UniCAR T cells can be retargeted to tumor cells via single-chain fragment variable (scFv)-based target modules (TMs). As schematically shown such scFv-based TMs represent a fusion molecule consisting of the respective scFv and the UniCAR epitope (E5B9). Thus, UniCAR T cells can form an immune complex with the TM, which allows the UniCAR T cell to recognize a specific target on the surface of the tumor cells. (b) As the development of a scFv-based TM for a clinical application is time-consuming and cost-extensive, we wanted to learn whether or not functional TMs can also be constructed from low-molecular-weight molecules: E.g. based on the PSMA-specific ligand PSMA-HBED-CC (PSMA-11 analogue). In order to transform this tracer in a potential TM, we fused the E5B9 epitope during chemical synthesis with PSMA-HBED-CC (see also ). The resulting compound PSMA peptide/ligand tracer TM (PSMA PLT-TM) cannot only be used for PSMA-PET imaging but also for retargeting of UniCAR T cells to PSMA-positive tumor cells.
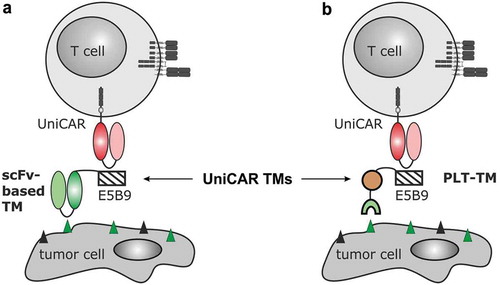
The aim of the presented study was to develop a novel theranostic PSMA ligand for management of PCa patients that combines the fields of nuclear medicine and CAR T cell technology. Thus, we converted PSMA-11, one of the most commonly used PCa imaging agent, by fusion of the 10-amino acid peptide epitope E5B9 into a TM for the UniCAR system (). The resulting chemically synthesized, low-molecular-weight compound, termed PSMA peptide/ligand tracer TM (PSMA PLT-TM) was functionally characterized with respect to its diagnostic as well as therapeutic properties in vitro and in vivo.
Results
Synthesis and radiolabeling of PSMA PLT-TM
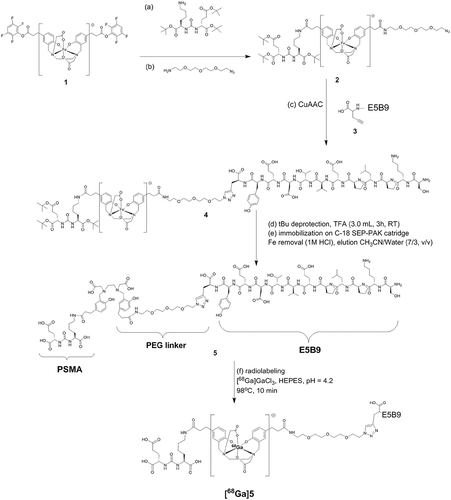
For theranostics of PCa, the novel low-molecular weight compound PSMA PLT-TM was chemically synthesized by fusion of the UniCAR epitope E5B9 to PSMA-11 (). The final product (see , product 5) was synthesized with purity greater than 98% (overall yield 15–20%), while radiolabeling resulted in one single species [68Ga]Ga-PSMA PLT-TM as determined by analytical reversed-phase high-performance liquid chromatography (RP-HPLC) with radiochemical purity ≥96% (Figure S1).
Figure 2. Synthesis of PSMA PLT-TM. The (tBu protected) PSMA-binding motif and a PEG linker were coupled to either side of the Fe-protected HBED-CC double TFP ester (1) to give (2). The synthesis of the C-terminally modified E5B9 peptide (SKPLPEVTDEY-Propargylglycine) (3) was performed manually on a 2CT-Resin under application of standard Fmoc-protocols and cleaved from it with a mixture of TFA/TIPS/H2O (95/2.5/2.5, v/v/v). Then, (2) and (3) reacted via the Cu(I)-catalyzed alkyne-azide cycloaddition (CuAAC), the final product was isolated and the tBu groups were deprotected with TFA. Then, Fe was removed from the complex by applying 1 M HCl on a C-18 SEP-PAK cartridge. The final product was eluted from the cartridge with a mixture of CH3CN/water (7/3, v/v, 20 mL).
Binding analysis of PSMA PLT-TM
First, an in vitro competitive cell-binding assay was performed for PSMA PLT-TM in order to determine its binding potential for the TAA PSMA in comparison with PSMA-11 using the PSMA-expressing LNCaP cell line. The results are expressed as percentage of cell-bound 68Ga-PSMA-10 in the presence of increasing concentrations of the non-labeled competitors PSMA PLT-TM and PSMA-11 (). PSMA PLT-TM presented a higher IC50 (50% inhibitory concentration) value (IC50 = 30.3 ± 1.1 nM) than the reference compound (PSMA-11, IC50 = 14.8 ± 1.2 nM).
Figure 3. Binding analysis of PSMA PLT-TM. (a) Displacement curves of 68Ga-PSMA-10 (30 nM) bound to PSMA expressed on LNCaP cells (105 cells per well). Results are expressed as % specific cell-bound radioactivity after incubation (45 min, RT) with increasing concentrations of non-radiolabeled PSMA-PLT TM or PSMA-11. The IC50 values are expressed as mean ± SD. Experiments were performed in quintuplicate. (b) 2 × 105 LNCaP or PC3 cells were incubated with 20 ng/µL TM. Binding was detected using the mouse anti-E5B9 and PE-labeled goat anti-mouse IgG Abs. In addition, cells were stained with mouse anti-human PSMA Ab/PE as positive control. Histograms show stained cells (blue line) and respective negative controls (black line). Percentage indicate proportion of PSMA+ cells under the marker. (c) For comparison of the binding affinity of the novel PSMA PLT-TM with the scFv-based αPSMA scFv-TM increasing amounts of the respective TM were incubated with LNCaP cells. The binding was estimated by flow cytometry. Relative median of fluorescence intensity (MFI) values were plotted against the concentration. Mean ± SEM of two different experiments is shown. KD values were calculated from the binding curves.
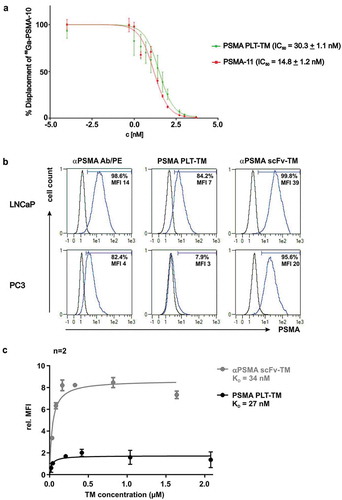
With regard to UniCAR T cell immunotherapy, we further verified that both binding sites of the bifunctional PSMA PLT-TM are accessible and capable to simultaneously interact with the respective partner domain (). Experiments were conducted in comparison to the previously described Ab-based αPSMA scFv-TM,Citation45,Citation54 which was purified from cell culture supernatants of eukaryotic cells using Nickel-NTA affinity chromatography (Figure S2). As shown by immunofluorescent staining of LNCaP cells, binding of both the PSMA PLT-TM and the αPSMA scFv-TM could be detected via the E5B9-tag (). Thus, the UniCAR epitope is still accessible for Ab binding which is the prerequisite for the interaction with UniCAR T cells. Using PC3 cells instead of LNCaP cells a binding of PSMA PLT-TM could be hardly detected (). As the staining of PC3 cells with both a commercial αPSMA mAb and the αPSMA scFv-TM also resulted in lower MFI values in comparison to LNCaP cells, this may be due to a low expression of PSMA on PC3 cells. Though the low expression level of PSMA on PC3 is still sufficient for retargeting of UniCAR T cells (as shown below) for technical reasons we selected the LNCaP cell line to estimate and compare KD values of the TM. For this purpose, increasing amounts of both TMs were incubated with LNCaP cells and the relative median of fluorescence intensity (MFI) values were determined by flow cytometry analysis as described previously.Citation45,Citation54 Based on the resulting binding curves (), we calculated KD values of 27 nM for the PSMA PLT-TM and 34 nM for the αPSMA scFv-TM. According to these data, PSMA PLT-TM and scFv-based αPSMA scFv-TM bind with similar affinity to PSMA.
Killing of PSMA-positive PCa cells by retargeted UniCAR T cells via PSMA PLT-TM occurs in a TM-dependent and target-specific manner with high efficacy similar to the scFV-based αPSMA scFV-TM
In order to evaluate the therapeutic potential of the novel PSMA PLT-TM, we first analyzed its capability to redirect UniCAR T cells for tumor cell killing. Therefore, human T cells from healthy donors were transduced with three lentiviral vectors encoding (I) the signaling construct UniCAR 28/ζ or (II) the UniCAR Stop construct without intracellular signaling domains or (III) the vector control expressing EGFP marker protein. The anti-tumor activity of redirected UniCAR T cells was analyzed by performing chromium release assays (see also Materials and methods) using both the high and low PSMA-expressing cell lines, LNCaP () and PC3 (). As shown in –b, both target cell lines were attacked by UniCAR 28/ζ T cells at effector-to-target cell (E:T) ratios between 5:1 and 1:5 only in the presence of TMs. Both, the PSMA PLT-TM and the scFv-based αPSMA scFv-TM worked comparably well. The retargeting of UniCAR T cells was TM dependent as tumor cell lysis was minimal and not statistically significant in the absence of TMs. As also expected, T cells modified with the UniCAR Stop vector (,b, UniCAR Stop) or the EGFP encoding vector control (,b, vector control) were neither in the presence nor in the absence of a TM able to induce significant tumor cell lysis. Using increasing amounts of the PSMA PLT-TM, we estimated EC50 (half-maximal effective concentration) values of 0.5 nM for LNCaP () and 9.2 nM for PC3 cells ().
Figure 4. Killing capability of PSMA PLT-TM redirected UniCAR T cells. (a–d) In order to compare the killing capability of UniCAR T cells (UniCAR 28/ζ) armed with either the PSMA PLT-TM or the αPSMA scFv-TM, chromium release assays were performed using either PSMA-positive (a, c) LNCaP or (b, d) PC3 tumor cells. (a, b) As negative controls served UniCAR T cells transduced with an EGFP-expressing vector control (vector control) or UniCAR construct lacking the signaling domains (UniCAR Stop). Data are reported as mean ± SEM for three independent donors. (***p < .0002, **p < .002, *p < .033 with respect to UniCAR 28/ζ w/o TM; 2way ANOVA with Tukey’s multiple comparisons test). (c, d) In order to estimate the range of working concentration for the PSMA PLT-TM PSMA-positive (c) LNCaP cells or (d) PC3 cells were co-cultivated with human T cells modified with the UniCAR signaling construct at an E:T ratio of 5:1. The TM was added at indicated concentrations. Specific lysis of tumor cells was estimated by chromium release assays (see Materials and methods). Data are reported as mean ± SEM for indicated number of independent donors. (***p < .0002, **p < .002 with respect to UniCAR 28/ζ w/o TM; 2way ANOVA with Sidak’s multiple comparisons test). (e) PSMA-positive PC3 cells were incubated with UniCAR T cells in the presence (w/TM) or absence (w/o TM) of the PSMA PLT-TM. Images were taken at the start (10 min), after 60 min, 120 min, 180 min, and 210 min.
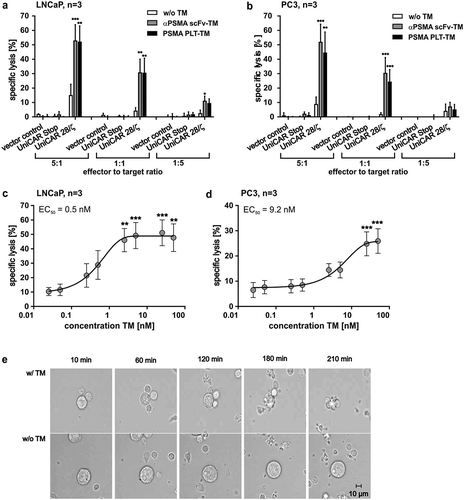
In order to visualize the time kinetic of tumor cell elimination by redirected UniCAR T cells in more detail, time-lapse microscopy was performed over 4 h. For this purpose, UniCAR T cells were incubated with LNCaP or PC3 cells in the presence or absence of the PSMA PLT-TM. As shown exemplarily for PC3 cells at selected time points, UniCAR T cells armed with the PSMA PLT-TM rapidly bind to and attack tumor cells (, w/TM). In the absence of the TM no stable interaction between UniCAR T cells and target cells were observed (, w/o TM). Similar results were obtained for LNCaP cells (data not shown).
After cross-linkage of PCa cells with UniCAR T cells via the PSMA PLT-TM cytokines are released in a TM- and target-specific manner
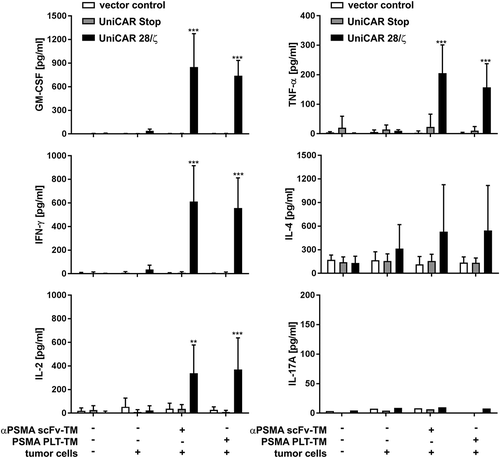
An important effector mechanism of redirected UniCAR T cells is the secretion of various pro-inflammatory and growth-promoting cytokines. Thus, we analyzed in a next step the capability of PSMA PLT-TM to induce cytokine secretion in redirected UniCAR T cells. For this purpose, T cells were genetically engineered to express either only EGFP (vector control), UniCAR Stop or UniCAR 28/ζ. Modified T cells were cultivated with or without PC3 or LNCaP tumor cells at an E:T ratio of 5:1 in the presence or absence of the scFv-based αPSMA scFv-TM or the PSMA PLT-TM for 24 h. As shown in Figure S3, cross-linkage of UniCAR T cells with both LNCaP and PC3 cells via the PSMA scFv-TM or the PSMA PLT-TM resulted in release of pro-inflammatory cytokines (TNF and IFN-γ). By using the MACSPlex Cytokine 12 Kit, human (see also Materials and methods), we could further show that only five of the 12 cytokines (GM-CSF, IFN-γ, IL-2, IL-4, and TNF-α) were detectable at relevant concentrations after retargeting of UniCAR T cells to PC3 cells via both TMs (). Again, these observations substantiate that cytokine secretion occurs in a strictly TM-dependent and target-specific manner. No significant difference between PSMA PLT-TM and αPSMA scFv-TM could be observed.
Figure 5. Analysis of cytokine release from redirected UniCAR T cells. Genetically engineered T cells (vector control, UniCAR Stop or UniCAR 28/ζ) were cultivated with or without PC3 cells (E:T = 5:1), either in the presence or in the absence of scFv-based αPSMA scFv-TM or PSMA PLT-TM. Data show concentrations of selected cytokines after 24 h and are reported as mean ± SD for four independent donors. (***p < .0002, **p < .002 with respect to UniCAR 28/ζ + tumor cells w/o TM; 2way ANOVA with Tukey’s multiple comparisons test).
PSMA PLT-TM engages UniCAR T cells for tumor cell killing in vivo
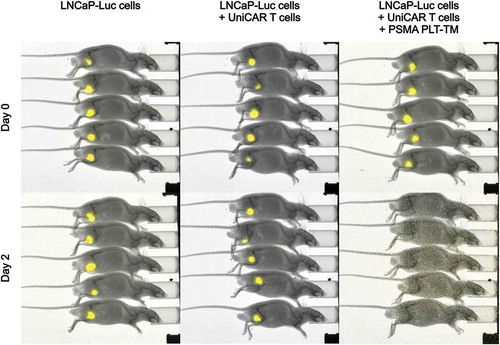
Recently, we have already successfully shown that the scFv-based αPSMA scFv-TM is able to redirect UniCAR T cells to eliminate PSMA-expressing tumor cells in experimental mice.Citation45 Now here, we analyzed whether the PSMA PLT-TM is also able to redirect UniCAR T cells for killing of PCa cells in vivo. To reduce the number of experimental mice, the in vivo experiments were limited to LNCaP cells transduced to overexpress firefly luciferase (termed LNCaP-Luc) as target cells (see Materials and methods). 1 × 106 LNCaP-Luc cells were mixed with 0.5 × 106 UniCAR 28/ζ T cells and 3 μM of PSMA PLT-TM per mouse (). As “untreated” controls served either 1 × 106 LNCaP-Luc cells alone or mixed with 0.5 × 106 UniCAR 28/ζ T cells without any TM. The respective mixtures (100 µL) were injected subcutaneously into female NMRI nu/nu mice resulting in three groups of animals each consisting of five mice. As shown in , and in agreement with the previously reported data for the scFv-based αPSMA scFv-TM,Citation45 already at day 2, no tumors were detectable in all animals of the treatment group while luciferase activity of tumor cells could be easily detected in all control mice. These data indicate that UniCAR T cells can also be armed in vivo with the PSMA PLT-TM for efficient tumor cell killing in a TM-dependent manner.
Figure 6. Retargeting of UniCAR T cells armed with the PSMA PLT-TM in experimental mice. 1 × 106 LNCaP-Luc tumor cells were mixed with 0.5 × 106 UniCAR 28/ζ T cells and 300 pmol/100 µL of the PSMA PLT-TM (LNCaP-Luc + UniCAR T cells + PSMA PLT-TM) per mouse. As “untreated” controls served either 1 × 106 LNCaP-Luc cells alone (LNCaP-Luc cells) or tumor cells were mixed with 0.5 × 106 UniCAR 28/ζ T cells without any TM (LNCaP-Luc + UniCAR T cells). The respective mixture (100 µL) was injected subcutaneously into female NMRI nu/nu mice resulting in three groups of animals each consisting of five mice. Luminescence imaging of anesthetized mice was performed 10 min after i.p. injection of 200 µL of luciferin (15 mg/mL) starting at day zero (day 0) and followed at day two (day 2).
PET imaging of PSMA-positive tumors using 68Ga-labeled PSMA PLT-TM
In order to show that the modified tracer PSMA PLT-TM can still be used for PET imaging, like PSMA-11, the PSMA PLT-TM was labeled with 68Ga (see Materials and methods). As shown in ,b, the modified PSMA tracer [68Ga]Ga-PSMA PLT-TM specifically enriched within the tumor of experimental mice. The binding of the radiolabeled PET tracer can be blocked with simultaneously injected unlabeled precursor PSMA PLT-TM that results in low molar activity of the radiotracer (,c). As expected the tracer is rapidly eliminated and therefore also found in the kidneys and in the bladder. A detailed biodistribution analysis of the tracer is presented in that confirmed the reduction of the 68Ga-activity concentration in the tumor by low molar radiotracer concentration. Moreover, administration of the competing drug 2-(phosphonomethyl)-pentandioic acid (2-PMPA) 50 min after radiotracer injection resulted in fast clearance of [68Ga]Ga-PSMA PLT-TM from the tumor indicating that [68Ga]Ga-PSMA PLT-TM specifically binds to PSMA ().
Table 1. Biodistribution of [68Ga]Ga-PSMA PLT-TM. The biodistribution of [68Ga]Ga-PSMA PLT-TM and of [68Ga]Ga-PSMA-11 was determined for three mice inoculated with LNCaP tumors, injected with approximately 10 MBq radiotracer and low (high molar activity) and high (low molar activity) amounts of PSMA PLT-TM. Values are provided as SUV (g/g). BW: body weight; BAT: brown adipose tissue; PSMA PLT-TM: PSMA peptide/ligand tracer target module; WAT: white adipose tissue; Subm.gl.: submandibular glands.
Figure 7. (a) PET imaging of the novel [68Ga]Ga-PSMA PLT-TM is shown as maximum intensity projections (MIP) and orthogonal planes (axial, coronal, sagittal) of two LNCaP tumor-bearing NMRI nu/nu mice. The distribution kinetics of [68Ga]Ga-PSMA PLT-TM (injected activity 15 MBq, molar activity 18 GBq/µmol, injected peptide amount 0.52 nmol PSMA PLT-TM) after single intravenous injection was measured in a dynamic PET study over 2 h and the images were prepared as integral activity from 60 to 120 min (midframe time 90 min). To demonstrate the specific binding of [68Ga]Ga-PSMA PLT-TM, PSMA was competitively blocked by simultaneous injection of 86 nmol/animal unlabeled PSMA PLT-TM in one animal (blocked). The green arrows show the tumor of the control animal (SUVmax 1.6 g/mL) and the red arrows indicate blocked tumors (SUVmax 0.06 g/mL). The color scales are in SUV (SUV, 0–1.6) and Hounsfield Units (HU, −1000–9093). (b) Blocking of [68Ga]Ga-PSMA PLT-TM tumor uptake. Estimation of (Bi) the blood and tumor uptake kinetics and (Bii) the corresponding tumor/blood and tumor/muscle ratios in the absence (control) or presence (blocked) of 86 nmol of unlabeled PSMA PLT-TM. Plots show mean ± SEM from experiments of four mice. (c) Kinetics of [68Ga]Ga-PSMA PLT-TM tumor uptake and blood activity concentration (SUVmean). Comparison of [68Ga]Ga-PSMA PLT-TM in LNCaP tumor-bearing mice without (control, n = 4) or with simultaneous injection of 86 nmol of non-radiolabeled PSMA PLT-TM (TM simultaneous, n = 4) and 50 min after radiotracer injection, injection of 2 µmol 2-PMPA (50 min p.i. PMPA, n = 2). The activity concentrations were derived from the ROIs over the tumors. Plots are presented as mean ± SEM of n experiments, respectively. (d) PET imaging of a patient with metastasized PCa comparing [68Ga]Ga-PSMA PLT-TM with [68Ga]Ga-PSMA-11. (Di) [68Ga]Ga-PSMA-11 was used for PET of a patient with metastasized PCa. (Dii) The same patient has undergone PET imaging 2 weeks later using [68Ga]Ga-PSMA PLT-TM. (Diii) PET scan of a healthy volunteer using [68Ga]Ga-PSMA PLT-TM.
![Figure 7. (a) PET imaging of the novel [68Ga]Ga-PSMA PLT-TM is shown as maximum intensity projections (MIP) and orthogonal planes (axial, coronal, sagittal) of two LNCaP tumor-bearing NMRI nu/nu mice. The distribution kinetics of [68Ga]Ga-PSMA PLT-TM (injected activity 15 MBq, molar activity 18 GBq/µmol, injected peptide amount 0.52 nmol PSMA PLT-TM) after single intravenous injection was measured in a dynamic PET study over 2 h and the images were prepared as integral activity from 60 to 120 min (midframe time 90 min). To demonstrate the specific binding of [68Ga]Ga-PSMA PLT-TM, PSMA was competitively blocked by simultaneous injection of 86 nmol/animal unlabeled PSMA PLT-TM in one animal (blocked). The green arrows show the tumor of the control animal (SUVmax 1.6 g/mL) and the red arrows indicate blocked tumors (SUVmax 0.06 g/mL). The color scales are in SUV (SUV, 0–1.6) and Hounsfield Units (HU, −1000–9093). (b) Blocking of [68Ga]Ga-PSMA PLT-TM tumor uptake. Estimation of (Bi) the blood and tumor uptake kinetics and (Bii) the corresponding tumor/blood and tumor/muscle ratios in the absence (control) or presence (blocked) of 86 nmol of unlabeled PSMA PLT-TM. Plots show mean ± SEM from experiments of four mice. (c) Kinetics of [68Ga]Ga-PSMA PLT-TM tumor uptake and blood activity concentration (SUVmean). Comparison of [68Ga]Ga-PSMA PLT-TM in LNCaP tumor-bearing mice without (control, n = 4) or with simultaneous injection of 86 nmol of non-radiolabeled PSMA PLT-TM (TM simultaneous, n = 4) and 50 min after radiotracer injection, injection of 2 µmol 2-PMPA (50 min p.i. PMPA, n = 2). The activity concentrations were derived from the ROIs over the tumors. Plots are presented as mean ± SEM of n experiments, respectively. (d) PET imaging of a patient with metastasized PCa comparing [68Ga]Ga-PSMA PLT-TM with [68Ga]Ga-PSMA-11. (Di) [68Ga]Ga-PSMA-11 was used for PET of a patient with metastasized PCa. (Dii) The same patient has undergone PET imaging 2 weeks later using [68Ga]Ga-PSMA PLT-TM. (Diii) PET scan of a healthy volunteer using [68Ga]Ga-PSMA PLT-TM.](/cms/asset/102e84d7-fc47-4268-a5a9-84558b43bca1/koni_a_1659095_f0007_oc.jpg)
In order to compare the diagnostic potential of the small molecule PSMA PLT-TM with the previously described αPSMA scFv-TM, we additionally labeled the scFv-based TM with 64Cu. PET imaging of LNCaP tumor-bearing mice revealed that maximum molar activity of [64Cu]Cu-NODAGA-αPSMA scFv-TM at the tumor site was detected after 24 h (Figure S4). Due to its small size and the extremely short half-life of the complexed radiometal 68Ga, maximum molar activity of [68Ga]Ga-PSMA PLT-TM could already be reached after 2 h (Figure S4B).
Finally, PET imaging results of the PSMA PLT-TM were compared with the original PET tracer PSMA-11 in order to further evaluate its potential as valuable tool for tumor diagnosis in humans. For this purpose, the 68Ga-labeled PSMA PLT-TM was used for imaging of a tumor patient with metastasized PCa (Figure 7Dii). Two weeks before, the same patient had undergone diagnostic examination via PET imaging using the conventional PET tracer [68Ga]Ga-PSMA-11 (Figure 7Di). As shown in , the 68Ga-labeled PSMA PLT-TM (Figure 7Dii) gives almost the same PET imaging result as obtained with the already clinically used [68Ga]Ga-PSMA-11 (Figure 7Di). Thus, modification of the PET tracer PSMA-11 with the UniCAR epitope had not dramatically changed the capability to use it as a PET tracer. The minor differences seen between the two PET scans of the patient (Figure 7Di-ii) may be caused by the treatment of the patient between the two scanning events but could possibly also be related to modifications made in the imaging agent. The PET scan of a healthy volunteer indicates tracer uptake into salivary glands (Figure 7Diii) which is in agreement with the expression of PSMA in this tissue. As monitored by PET scans of the tumor patient, not only [68Ga]Ga-PSMA PLT-TM but also the conventional tracer [68Ga]Ga-PSMA-11 is able to accumulate in this gland tissue (Figure 7Di-ii).
Discussion
So far, theranostics in the context of PCa is based on nuclear medicine, namely the combination of small molecules or Abs for PET imaging and radioligand therapy.Citation5,Citation6,Citation16,Citation17,Citation22 However, the here described PSMA PTL-TM goes beyond this strategy and represents for the first time a novel immunotheranostic agent that allows both PCa imaging and T cell-based immunotherapy. For proof of concept, we selected the clinically used Glu-ureido-based PSMA-binding motif Glu-urea-Lys linked to the chelator HBED-CC. During chemical synthesis of the novel, PSMA PLT-TM tracer 11 amino acids representing the UniCAR epitope and additionally propargylglycine for the C-terminal conjugation of the peptide were added to the PSMA-11 analogue by CuAAC to the mini-PEG linker attached to second propionic acid function of the chelator. Our data provide experimental evidence in mice and humans that PSMA-11 could be successfully converted into a UniCAR TM without affecting its use as tracer for PSMA-PET imaging.
From an immunotherapeutic point of view, the PSMA PLT-TM was highly effective as TM for retargeting of UniCAR T cells to kill PSMA-positive PCa cells both in vitro and in vivo. Thereby, UniCAR T cell efficacy and cytokine profile was comparable to the already established αPSMA scFv-TM. Low numbers of UniCAR T cells as well as low concentrations of PSMA PLT-TM were sufficient to engage UniCAR T cells for tumor cell lysis. After 24 h, already the majority of tumor cells was eliminated in vitro. In principle, redirected UniCAR T cells should be able to eradicate all target cells over time as CAR T cells are serial killers.Citation55 This is in line with our in vivo experiment showing complete tumor destruction already after 2 days. Moreover, with EC50 values in a low nM range, killing efficiency of UniCAR T cells armed via the PSMA PLT-TM is comparable to all so far described combinations with Ab-based TMs.Citation44–Citation50 Furthermore, we have shown for the first time that not only Ab fragments but also peptide ligands can be successfully fused to the UniCAR epitope E5B9. In context of solid tumor UniCAR T cell therapy, such small molecules are advantageous due to increased tumor permeability and short serum half-lifes.Citation16 From an economic point of view, utilization of a PLT-TM is very favorable for drug development as GMP production is less cost-intensive and less time-consuming in comparison to Ab-based TMs. In a clinical setting, the UniCAR system seems to be a promising approach as it maintains high anti-tumor activity of conventional CARs while providing an additional safety switch. After intravenous injection of the UniCAR T cell product, the patient will receive continuous infusions of appropriate TM doses. For convenient treatment of PCa patients, it would be possible to administer first low doses of the rapidly eliminated PSMA PLT-TM until the majority of tumor cells have been eliminated. This allows a prompt intervention (stopping TM infusion) until the risk of severe acute side effects including tumor lysis syndrome and/or cytokine release syndrome becomes low due to reduced tumor burden. As shown by blocking experiments in experimental mice (see ), administration of the competing agent 2-PMPA could also be used as an emergency medicine to rapidly eliminate TMs bound to PSMA-expressing tissues. During the course of treatment, the TM dose can be subsequently increased and adjusted in dependence on the occurrence of adverse reactions. Also, TMs with longer half-life or even a cell factory producing an antibody-based TM in vivo may be applied instead of a continuous TM infusion.Citation48 In order to reduce the risk of tumor escape variants, a simultaneous application of two or even more TMs would be possible.Citation44,Citation45 If tumor cells up-regulate or already express resistant factors such as checkpoint inhibitors, TMs co-delivering stimulatory signals (as already shown for the modular bispecific T cell engager format [e.g. 52]) could help to circumvent this problem. Moreover, UniCAR T cell therapy can be shut down after molecular remission to avoid long-term destruction of healthy tissues, e.g. salivary glands and kidneys showing low PSMA expression. In case of tumor relapse, UniCAR T cells can be easily reactivated by TM infusion including to a different target by switching TM specificity.
With respect to its role as a potential PET radioligand, we have shown that the novel PSMA PLT-TM can be complexed rapidly and efficiently with 68Ga. The resulting [68Ga] 68Ga-PSMA PLT-TM was characterized by high purity underlining its suitability as PET radiotracer. It is well known that already small changes in the chemical structure of Glu-ureido-based PSMA ligands, can affect PSMA binding and thus their imaging properties.Citation16 We could prove that after modification of PSMA-11 with the peptide E5B9 affinity toward PSMA could be maintained and is still in the nM range (IC50 value of 30.3 ± 1.1 nM). As required for high image quality in PCa patients, PSMA PLT-TM was also able to rapidly and specifically accumulate at the tumor site both in tumor-bearing mice and in one PCa patient. By performing a first in human study, we could show that it allows the detection of different tumor lesions in men with low imaging background similar to PSMA-11. Thereby, visualization of bone and lymph node metastases is especially important for high-risk PCa patients. Having used the PSMA PLT-TM for imaging of just one patient, these data are limited and need to be confirmed by imaging of further patients at best in the context of the planned phase 1 UniCAR trial. Nonetheless, the PSMA PLT-TM is a promising theranostic agent since we observed only minor differences of human PET/CT scans between [68Ga]Ga-PSMA-11 and [68Ga]Ga-PSMA PLT-TM which may be explained by the fact that the patient was treated between the two measurements. Of course we currently cannot exclude that the observed minor differences, e.g. the more intense staining of the kidneys are caused by alterations in the chemical structure of the tracer molecule. Due to its small size and the short half-life of the radiometal 68Ga (68 min), the diagnostic window of [68Ga]Ga-PSMA PLT-TM seems to be suitable for fast patient examinations directly after radiotracer injections but might also be limited to a few hours. Thus, utilization of the 64Cu-labeled αPSMA scFv-TM represents one possibility to extend the diagnostic range of imaging up to 2 days after injection.
Like PSMA-11, the novel PSMA PLT-TM accumulated as expected also in non-cancerous organs, e.g. kidneys and salivary glands which are known to express lower PSMA levels. However, based on the herein presented data it is assumed that the PSMA-11-derived PSMA PLT-TM is still a reliable imaging and immunotherapeutic agent. In case severe side effects against healthy tissues occur, the PSMA PLT-TM can be rapidly removed from the body by both stopping TM supply and by administration of the competing agent 2-PMPA as already mentioned above.
Overall, the results indicate that the PSMA PLT-TM is an attractive candidate for an immunotheranostics in PCa patients. The radiolabeled compound might be a useful tool for initial diagnosis as well as for staging and monitoring of tumor disease during and after therapy. The unlabeled counterpart is instead intended as a TM for cancer immunotherapy with UniCAR T cells. Thereby, the radiolabeled PSMA PLT-TM can be used periodically for target verification and assessment of molecular response to therapy. To allow high-quality imaging and to avoid blockade of PSMA by unlabeled PSMA PLT-TM, TM infusions can briefly be stopped before performing [68Ga]Ga-PSMA PLT-TM PET imaging. In general, such a theranostic approach paves the way for personalized medicine in PCa and is especially important if PSMA-negative tumor escape variants occurs. It will therefore definitely help in decision-making and can guide an appropriate patient-specific treatment: e.g. switching to alternative TMs if TAA-negative tumor escape variants develop or stopping UniCAR T cell therapy in case severe side effects against healthy tissue occur. Local radiation of solid tumor lesions during PET imaging might also be supportive for UniCAR T cell immunotherapy as ionizing radiation can increase CAR T cell infiltrations, for example, by triggering the up-regulation of certain tumor antigens, adhesion molecules and the release of T cell attracting chemokines.Citation56,Citation57 In addition, an increased PSMA expression following androgen deprivation therapy was reportedCitation58 and is thus an attractive starting point for further therapy combinations with PSMA PLT-TM theranostics.
Summing up, in combination with UniCAR T cells, the PSMA PLT-TM is a highly promising theranostic compound that might be useful for retargeting of UniCAR T cells to PCa tissues and for following the progress of tumor treatment via PET imaging. Moreover, other small molecule-based ligands binding to overexpressed tumor surface molecules may also be converted into such immunotheranostic compounds.
Materials and methods
Cell lines
Recombinantly or natively PSMA-expressing PC3 or LNCaP C4-2B PCa cells (termed here PC3 or LNCaP) were used as target cell lines and cultured in RPMI 1640 complete medium (Biochrom, Berlin, Germany) as described previously.Citation45,Citation59 3T3 cells modified to express the scFv-based αPSMA scFv-TM were kept in DMEM complete medium (Invitrogen, ThermoFisher Scientific, Schwerte, Germany).Citation59 Incubation of all cell lines was done at 37°C and 5% CO2.
Synthesis of PSMA PLT-TM
Commercially available analytical grade chemicals were used and not further purified. The chemical suppliers were: Sigma-Aldrich (Taufkirchen, Germany) and Merck (Darmstadt, Germany), unless otherwise indicated. Protected amino acids and resins were supplied from Novabiochem (Merck, Darmstadt, Germany) and IRIS Biotech (Marktredwitz, Germany).
The following RP-HPLC systems were used for analysis and purification: Agilent 1100 Series, equipped with a multi-wavelength-detector and Latek P402 (Latek Labortechnik-Geraete, Eppelheim, Germany) equipped with a HITACHI variable UV detector (absorbance measurement at 214 and 254 nm) and a gamma detector (Bioscan, Washington, USA). Both RP-HPLC systems were equipped with either an analytical Chromolith RP-18e (4.6 × 100 mm; Merck, Darmstadt, Germany) for analysis or a semi-preparative column Chromolith RP-18e (10 × 100 mm; Merck, Darmstadt, Germany) for purifications.
The MALDI-MS Daltonics Microflex (Bruker Daltonics, Bremen, Germany) system was used for mass spectrometry. Full-scan single mass spectra were obtained by scanning m/z = 400–4000.
Synthesis of PSMA PLT-TM is summarized in and described in detail in the following chapters:
Synthesis of 1 and 2
The synthesis of the bis (TFP)ester of N,N´-bis-[2-hydroxy-5(carboxyethyl)benzyl] ethylenediamine-N,N´- diacetic acid, (1), [Fe(HBED-CC)]TFP2 iron complex was performed as described previously.Citation60,Citation61 Fifty-six milligrams of [Fe(HBED-CC)]TFP2 (0.095 mmol) was dissolved on 300 µL of dry DMF (Sigma-Aldrich, Taufkirchen, Germany). After addition of 20 µL of DIPEA (1.2 equiv.) and 41.8 mg of di-tert-butyl (((S)-6-amino-1-(tert-butoxy)-1-oxohexan-2-yl)carbamoyl)-L-glutamate (ABX, Radeberg, Germany) (0.9 equiv.) the solution was stirred for 5 h at room temperature. Finally, 50 µL of 11-Azido-3,6,9-trioxaundecan-1-amine (Sigma-Aldrich, Taufkirchen, Germany) (2.6 equiv.) was added and the mixture was stirred for 48 h at room temperature. The product (2) was isolated via RP-HPLC by using a linear gradient (A-B): 20–65% B in 16.2 min (Rt: 14.1 min); A: 0.1% TFA in H2O and B: 0.1% TFA in CH3CN, flow rate 5 mL/min. Maldi-MS (m/z) for [M + H]+ (calculated for C58H91N9O18): 1202.4 (1201.65).
Synthesis of C-terminally modified peptide (3)
The synthesis of the C-terminally modified UniCAR peptide (SKPLPEVTDEY-Propargylglycine) (3) was performed manually on a 2CT-Resin (Merck, Darmstadt, Germany) in a 0.15 mmol scale under application of standard Fmoc-protocols. After cleavage of the peptide from the resin with a mixture of TFA/TIS/Water (95/2.5/2.5, v/v/v) the peptide was purified via RP-HPLC with a linear gradient (A-B): 10–37% B in 12.3 min; A: 0.1% TFA in H2O and B: 0.1% TFA in CH3CN, flow rate 5 mL/min, Rt: 11.8 min. After lyophilization 30 mg of pure product were obtained (yield: 12.5%), Maldi-MS (m/z) for [M + H]+ (calculated for C62H93N13O22): 1371.7 (1371.66).
Synthesis of 4 and 5
The coupling of the alkyne bearing peptide (3) with the PSMA-ligand (intermediate 2) was performed via the Cu(I)-catalyzed alkyne-azide cycloaddition (CuAAC). 7.74 mg of 2 (0.0062 mmol) were dissolved together with 13.4 mg of peptide 3 (0.0098 mg; 1.4 equiv.) in 500 µL THF/H2O. Then, 4.1 mg of CuSO4 (0.0248 mmol, 4 equiv.) and 4.9 mg of sodium ascorbate (0.0248 mmol, 4 equiv.) were added and the mixture was left to stir for 16 h (RT). After purification by means of RP-HPLC the intermediate tris(tBu)-protected intermediate (4) was isolated and lyophilized. Gradient applied (A-B): 15–54% B in 14.2 min, flow: 5 mL/min, A: 0.1% TFA in H2O and B: 0.1% TFA in CH3CN, Rt: 13.4 min, Maldi-MS (m/z) [M + H]+ (calculated for C120H184N22O40) 2574.3 (2573.30). After cleaving the tert.-butyl-protecting groups from the PSMA-binding motif with TFA (3 mL, 3 h, RT) TFA was evaporated. The iron containing intermediate was dissolved in 2% CH3CN in water (50 mL) and immobilized on a C18-SEP-PAK Cartridge (Waters, Eschborn, Germany). The iron was removed by eluting the C-18 cartridge with 10 mL of 1 M HCl. After washing with water the product 5 (PSMA PLT-TM) was eluted with CH3CN/Water (7/3, v/v, 20 mL). After lyophilization 10 mg of the product were obtained as a white powder (yield 60%), Maldi-MS (m/z) for [M + H]+ (calculated for C108H160N22O40): 2406.5 (2405.12).
Radiolabeling of the final product with 68Ga was accomplished according to previously published methods for 68Ga-PSMA-11.Citation60,Citation61 Briefly, a solution of product 5 (1.0 nmol in 0.1 M HEPES buffer, pH 7.5, 100 μL) was added to a mixture of 68[Ga]GaCl3 (80–100 MBq, 40 μL) eluate and HEPES (2.1 M, 10 μL), at pH 4.2 (98°C, 10 min). Radiochemical purity was analyzed by RP-HPLC (Zorbax C18 300SB 9.4 x 250, 4 µm) and gradient H2O (1)/MeCN (2) containing 0.1% TFA with 5 min to 95% (1), during 10 min to 95% (2), 5 min to 95% (2) and recovery of the system in 5 min to 95% (1) and 5 min to 95% (1). The flow rate was 3 mL/min.
Determination of binding affinity for PSMA in LNCaP cells
Binding affinities (IC50) for the compounds under study were determined by a competitive cell-binding assay with PSMA-positive LNCaP cells, using known protocols.Citation60,Citation61 On a 96-well plate (MultiScreen HTS-DV 0.65 μm) LNCaP cells (105 per well) were incubated with 0.75 nM [68Ga]Ga-HBED-CC-[Glu-urea-Lys(Ahx)]2 (68Ga-PSMA-10) (16–20 MBq/nmol) in the presence of 12 different concentrations of PSMA PLT-TM or PSMA-11 ranging from 0 − 5000 nM (100 μL/well). After incubation at RT for 45 min with gentle agitation, the binding buffer was removed using a multiscreen vacuum manifold (Millipore, Billerica, MA). After washing twice with 100 μL and once with 200 μL ice-cold binding buffer, the cell-bound radioactivity was measured with a gamma counter. The 50% inhibitory concentration (IC50) and values were calculated by fitting the data using a nonlinear regression algorithm (GraphPad Software, La Jolla, CA, USA). Experiments were performed in quintuplicate.
Purification of recombinant antibodies
The scFv-based anti-PSMA TM (here termed as αPSMA scFv-TM) was produced by a stable 3T3 cell line.Citation54 For purification of recombinant Abs from cell culture supernatants, Ni-NTA affinity chromatography was performed.Citation54,Citation62,Citation63 In order to evaluate purity and concentration of the αPSMA, scFv-TM SDS-PAGE and immunoblotting were done as already described.Citation54,Citation59
Generation of UniCAR vectors
Cloning of the UniCAR 28/ζ vector as well as of the vector control encoding the EGFP marker protein and the UniCAR Stop construct lacking intracellular signaling domains has been described in detail elsewhere.Citation33,Citation44,Citation52,Citation64,Citation65
Isolation of peripheral blood mononuclear cells and T cell subpopulations
Isolation and cultivation of primary human T cells was done as already published.Citation45,Citation59 For this purpose, either buffy coats (supplied by German Red Cross, Dresden, Germany) or fresh blood of healthy donors was utilized.Citation59 The study including the consent form was approved by the local ethics committee of the Medical Faculty of the University Hospital Carl Gustav Carus, TU Dresden (EK27022006).
Lentiviral transduction of T cells
Transduction of human T cells with lentiviral particles was conducted as described previously [e.g. 33, 45, 49]. In order to ensure that all assays are performed with T cell populations consisting of >90% UniCAR 28/ζ T cells (UniCAR Stop T cells/vector control T cells), they were sorted using a BD FACSAria Fusion (BD Biosciences Pharmingen, Heidelberg, Germany). Genetically modified T cells were cultured in RPMI 1640 complete media supplemented with cytokines IL-2 (Proleukin®, Novartis Pharmaceuticals, #2238131), IL-7 (ImmunoTools, #11340075) and IL-15 (ImmunoTools, #11340155).Citation33,Citation45,Citation49 Media was replaced by RPMI 1640 complete media lacking any recombinant cytokines 24 h before experiments started.Citation33,Citation45,Citation49
Cytokine release assay
In a cytokine release assay, 2.5 × 10Citation4 UniCAR T cells were incubated with or without 5 × 103 PC3 or LNCaP cells in the presence or absence of the respective TM for 24 h. After centrifugation (360xg, 5 min), cell-free supernatants were collected. Cytokines were subsequently quantified by using the MACSPlex Cytokine 12 Kit, human (Miltenyi Biotec GmbH, #130–099-169) or the OptEIATM Human TNF (BD Biosciences, #555212) and OptEIATM Human IFN-γ ELISA Kits (BD Biosciences, #555142).Citation45,Citation46
Flow cytometry analysis
For TM binding analysis, tumor cells were firstly stained with different concentrations of TM, secondly a mouse anti-E5B9 mAb directed against the epitope E5B9 of the TM, and thirdly a PE-conjugated goat F(ab’)2 fragment anti-mouse IgG (Fcg)-PE (Beckmann Colter,#IM0551) detection Ab. The directly labeled mouse anti-human PSMA Ab/PE (MBL International, #K0142-5) served as positive control. Stained cells were analyzed using a MACSQuant® Analyzer and MACSQuantify® software (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). Relative median of fluorescence intensities of the stained cells were used to create a binding curve and calculate the respective KD value with the GraphPad Prism software (GraphPad Software Inc., La Jolla, CA, USA) [e.g. 46].
Cytotoxicity assay
To analyze UniCAR T cell-mediated tumor cell killing standard chromium release assays were performed.Citation66 For this purpose, genetically modified effector T cells were co-cultivated with 5 × 103 51Cr-labeled target cells (PSMA-expressing LNCaP or PC3 cells) in the presence or absence of either the scFv-based αPSMA scFv-TM or the PSMA PLT-TM at indicated E:T ratios. After 24 h or 48 h, specific lysis of tumor cells was determined as described previously.Citation66
Time-lapse microscopy
For live imaging, PC3 or LNCaP tumor cells were mixed with UniCAR T cells at an E:T ratio of 5:1 without TM or with 30 nM of the PSMA PLT-TM. Cells were cultured in RPMI complete media in an eight well chamber slide (ibidi, Martinsried, Germany). Imaging was performed over 4 h in a humidified atmosphere at 37°C with the all-in-one fluorescence microscope BZ-X700 (Keyence, Japan).
Immuno-pet imaging, biodistribution analysis, and optical imaging of tumor xenograft models
All animal experiments were carried out at the Helmholtz-Zentrum Dresden-Rossendorf (HZDR) according to the guidelines of German Regulations for Animal Welfare and have been approved by the Landesdirektion Dresden (24–9165.40–4, 24.9168.21–4/2004–1).
Immuno-PET imaging and biodistribution analysis was carried out in 5–8 week old naive, athymic male nude NMRI-nu Rj:NMRI-Foxn1nu/nu mice (Janvier Labs, France) (named NMRI nu/nu) as previously published.Citation46,Citation47 For this purpose, 1 × 106 tumor cells (in PBS) were inoculated subcutaneously in the right hind flank or the right shoulder. Once tumors reached a size of 100–500 mm3, PET imaging or biodistribution analysis were performed. Specific tumor enrichment of 68Ga-labeled PSMA PLT-TM radiotracer was blocked by high doses of unlabeled PSMA PLT-TM. Alternatively, specifically bound PSMA PLT-TM radiotracer was removed by intravenous administration of the competing drug 2-PMPA 50 min after radiotracer injection.
For in vivo analysis of tumor cell killing by TM-redirected UniCAR T cells, 1 × 106 LNCaP-Luc cells were mixed with 0.5 × 106 human UniCAR 28/ζ T cells in the presence or absence of the PSMA PLT-TM (5 mice per group). As control, one experimental group (n = 5) received tumor cells alone. The cell mixtures (100 µL) were subcutaneously injected into the right thigh of experimental mice. Luminescence imaging (exposure times 1 s, 10 s, and 60 s) was performed as previously described.Citation45–Citation50
PET imaging of a human patient with metastasized PCa
Radiolabeling of PSMA PLT-TM: For PET imaging of a patient with metastasized PCa PSMA-11 and PSMA PLT-TM were labeled with 68Ga. Labeling of 30 µg PSMA PLT-TM and 20 µg PSMA-11 with 68Ga was done similar to other peptides as previously described for [68Ga]Ga-DOTA-TATE.Citation67 Samples were taken for determination of identity, radiochemical purity and pH.
Imaging: [68Ga]Ga-PSMA-11 and [68Ga]Ga-PSMA PLT-TM PET/computed tomography (CT) scans were performed in a man with multiple metastases of PCa with 3-months interval with radionuclide therapy in between. Images were acquired 60 min after injection of 170 MBq [68Ga]Ga-PSMA-11 and 65 min after injection of 164 MBq [68Ga]Ga-PSMA PLT-TM. PET/CT scans were performed from the base of the skull to mid-thigh on a Biograph 16 HI-REZ scanner (Siemens/CTI, Knoxville, TN). Seven bed positions of 3 min each (axial field of view 15 cm, overlap 1 cm) were acquired. CT imaging was carried out as a low-dose scan without contrast enhancement (120 kV, 10 mAs, 512 × 512 matrix) for attenuation correction only. Images were reconstructed using the ordered subsets expectation maximization algorithm using six iterations and four subsets and CT-based attenuation correction. The study and consent were approved by the local ethics committee of the Technical University (TU) Dresden (EK491122018). A written informed consent was obtained from the patient as well as from the healthy volunteer (male).
Statistical analysis
Statistical analysis was performed with GraphPad Prism software version 7 (GraphPad Software Inc., La Jolla, CA, USA) as indicated in the figure legends.
Author contributions
CA, AF, SK, MS, DB, AK, JAS, EPC, NM performed experiments and analyzed the data. M. Schäfer synthesized the PSMA PLT-TM. RB and NB performed in vivo studies in experimental mice including the PET imaging. GW and JK performed the human PET analyses. JP, M. Schmitz, GE, KK, CL, M. Schäfer and MB provided critical reagents, know how, and materials. MB, SK, CA, and AF conceived and designed the experiments. MB had the idea to convert the PSMA-PET tracer into a TM, supervised the experiments, and wrote the manuscript.
Disclosure of Potential Conflicts of Interest
MB and GE have filed patents related to the UniCAR system. MB and GE are shareholders of the company GEMoaB which owns the IP related to the UniCAR system. GE is owner of the company Cellex PT. The other authors have declared that no competing interest exists.
Supplemental Material
Download Zip (397.4 KB)Acknowledgments
We thank Julia Lagler, Regina Herrlich and Andrea Suhr for excellent technical assistance.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Related Research Data
References
- Drake CG. Prostate cancer as a model for tumour immunotherapy. Nat Rev Immunol. 2010;10:1–15. doi:10.1038/nri2817.
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86. doi:10.1002/ijc.29210.
- Walsh PC, DeWeese TL, Eisenberger MA. Clinical practice. Localized prostate cancer. N Engl J Med. 2007;357:2696–2705. doi:10.1056/NEJMcp0706784.
- Pound CR, Partin AW, Eisenberger MA, Chan DW, Pearson JD, Walsh PC. Natural history of progression after PSA elevation following radical prostatectomy. JAMA. 1999;281:1591–1597. doi:10.1001/jama.281.17.1591.
- Virgolini I, Decristoforo C, Haug A, Fanti S, Uprimny C. Current status of theranostics in prostate cancer. Eur J Nucl Med Mol Imaging. 2018;45:471–495.
- Kulkarni HR, Singh A, Langbein T, Schuchardt C, Mueller D, Zhang J, Lehmann C, Baum RP Theranostics of prostate cancer: from molecular imaging to precision molecular radiotherapy targeting the prostate specific membrane antigen. Br J Radiol. 2018;91:20180308.
- Kawakami M, Nakayama J. Enhanced expression of prostate-specific membrane antigen gene in prostate cancer as revealed by in situ hybridization. Cancer Res. 1997;57:2321–2324.
- Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res. 1997;3:81–85.
- Mhawech-Fauceglia P, Zhang S, Terracciano L, Sauter G, Chadhuri A, Herrmann FR, Penetrante R. Prostate-specific membrane antigen (PSMA) protein expression in normal and neoplastic tissues and its sensitivity and specificity in prostate adenocarcinoma: an immunohistochemical study using multiple tumour tissue microarray technique. Histopathology. 2007;50:472–483.
- Kiessling A, Wehner R, Füssel S, Bachmann M, Wirth MP, Schmitz M. Tumor-associated antigens for specific immunotherapy of prostate cancer. Cancers (Basel). 2012;4:193–217. doi:10.3390/cancers4010193.
- Cunha AC, Weigle B, Kiessling A, Bachmann M, Rieber EP. Tissue-specificity of prostate specific antigens: comparative analysis of transcript levels in prostate and non-prostatic tissues. Cancer Lett. 2006;236:229–238. doi:10.1016/j.canlet.2005.05.021.
- Raff AB, Gray A, Kast WM. Prostate stem cell antigen: a prospective therapeutic and diagnostic target. Cancer Lett. 2009;277:126–132. doi:10.1016/j.canlet.2008.08.034.
- Ross JS, Sheehan CE, Fisher HA, Kaufman RP Jr, Kaur P, Gray K, Webb I, Gray GS, Mosher R, et al. Correlation of primary tumor prostate-specific membrane antigen expression with disease recurrence in prostate cancer. Clin Cancer Res. 2003;9:6357–6362.
- Afshar-Oromieh A, Haberkorn U, Eder M, Eisenhut M, Zechmann CM. [68Ga]Gallium-labelled PSMA ligand as superior PET tracer for the diagnosis of prostate cancer: comparison with 18F-FECH. Eur J Nucl Med Mol Imaging. 2012;39:1085–1086. doi:10.1007/s00259-012-2069-0.
- Afshar-Oromieh A, Malcher A, Eder M, Eisenhut M, Linhart HG, Hadaschik BA, Holland-Letz T, Giesel FL, Kratochwil C, Haufe S, et al. PET imaging with a [68Ga]gallium-labelled PSMA ligand for the diagnosis of prostate cancer: biodistribution in humans and first evaluation of tumour lesions. Eur J Nucl Med Mol Imaging. 2013;40:486–495. doi:10.1007/s00259-012-2298-2.
- Kopka K, Benešová M, Bařinka C, Haberkorn U, Babich J. Glu-ureido-based inhibitors of prostate-specific membrane antigen: lessons learned during the development of a novel class of low-molecular-weight theranostic radiotracers. J Nucl Med. 2017;58:17S–26S. doi:10.2967/jnumed.116.186775.
- Cuccurullo V, Di Stasio GD, Mansi L. Nuclear medicine in prostate cancer: a new Era for radiotracers. World J Nucl Med. 2018;17:70–78. doi:10.4103/wjnm.WJNM_54_17.
- Eder M, Schäfer M, Bauder-Wüst U, Hull WE, Wängler C, Mier W, Haberkorn U, Eisenhut M. 68Ga-complex lipophilicity and the targeting property of a urea-based PSMA inhibitor for PET imaging. Bioconjug Chem. 2012;23:688–697. doi:10.1021/bc200279b.
- Benešová M, Bauder-Wüst U, Schäfer M, Klika KD, Mier W, Haberkorn U, Kopka K, Eder M. Linker modification strategies to control the Prostate-Specific Membrane Antigen (PSMA)-Targeting and Pharmacokinetic properties of DOTA-conjugated PSMA Inhibitors. J Med Chem. 2016;59:1761–1775. doi:10.1021/acs.jmedchem.5b01210.
- Benešová M, Schäfer M, Bauder-Wüst U, Afshar-Oromieh A, Kratochwil C, Mier W, Haberkorn U, Kopka K, Eder M. Preclinical evaluation of a tailor-made DOTA-conjugated PSMA inhibitor with optimized linker moiety for imaging and endoradiotherapy of prostate cancer. J Nucl Med. 2015;56:914–920. doi:10.2967/jnumed.114.147413.
- Weineisen M, Schottelius M, Simecek J, Baum RP, Yildiz A, Beykan S, Kulkarni HR, Lassmann M, Klette I, Eiber M, et al. 68Ga- and 177Lu-labeled PSMA I&T: optimization of a PSMA-targeted theranostic concept and first proof-of-concept human studies. J Nucl Med. 2015;56:1169–1176. doi:10.2967/jnumed.115.158550.
- Rahbar K, Afshar-Oromieh A, Jadvar H, Ahmadzadehfar H. PSMA theranostics: current status and future directions. Mol Imaging. 2018;17:1536012118776068. doi:10.1177/1536012118776068.
- Abken H. Adoptive therapy with CAR redirected T cells: the challenges in targeting solid tumors. Immunotherapy. 2015;7:535–544. doi:10.2217/imt.15.15.
- Stauss HJ, Morris EC, Abken H. Cancer gene therapy with T cell receptors and chimeric antigen receptors. Curr Opin Pharmacol. 2015;24:113–118. doi:10.1016/j.coph.2015.08.006.
- Holzinger A, Barden M, Abken H. The growing world of CAR T cell trials: a systematic review. Cancer Immunol Immunother. 2016;65:1433–1450. doi:10.1007/s00262-016-1895-5.
- Cartellieri M, Bachmann M, Feldmann A, Bippes C, Stamova S, Wehner R, Temme A, Schmitz M. Chimeric antigen receptor-engineered T cells for immunotherapy of cancer. J Biomed Biotechnol. 2010; 2010:956304.
- Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, Lewis ID, Brenner MK, Brown MP. GD2-specific CAR T Cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol Ther. 2016;24:1135–1149. doi:10.1038/mt.2016.63.
- Arcangeli S, Rotiroti MC, Bardelli M, Simonelli L, Magnani CF, Biondi A, Biagi E, Tettamanti S, Varani L. Balance of Anti-CD123 chimeric antigen receptor binding affinity and density for the targeting of acute Myeloid Leukemia. Mol Ther. 2017;25:1933–1945. doi:10.1016/j.ymthe.2017.04.017.
- Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, Jones DR, Sadelain M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;261:261ra151. doi:10.1126/scitranslmed.3010162.
- Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJ, Scholler J, Song D, Porter DL, Carroll M. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29:1637–1647. doi:10.1038/leu.2015.52.
- Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell. 2017;168:724–740. doi:10.1016/j.cell.2017.01.016.
- Sadelain M. CAR therapy: the CD19 paradigm. J Clin Invest. 2015;125:3392–3400. doi:10.1172/JCI80010.
- Cartellieri M, Koristka S, Arndt C, Feldmann A, Stamova S, von Bonin M, Töpfer K, Krüger T, Geib M, Michalk I, et al. A novel ex vivo isolation and expansion procedure for chimeric antigen receptor engrafted human T cells. PLoS One. 2014;9:e93745. doi:10.1371/journal.pone.0093745.
- Zuccolotto G, Fracasso G, Merlo A, Montagner IM, Rondina M, Bobisse S, Figini M, Cingarlini S, Colombatti M, Zanovello P, et al. PSMA-specific CAR-engineered T cells eradicate disseminated prostate cancer in preclinical models. PLoS One. 2014;9:e109427. doi:10.1371/journal.pone.0109427.
- Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75. doi:10.1038/nbt.2459.
- Miller BC, Maus MV. CD19-targeted CAR T cells: a new tool in the fight against B cell malignancies. Oncol Res Treat. 2015;38:683–690. doi:10.1159/000442170.
- Davila ML, Brentjens RJ. CD19-Targeted CAR T cells as novel cancer immunotherapy for relapsed or refractory B-cell acute lymphoblastic leukemia. Clin Adv Hematol Oncol. 2016;14:802–808.
- Zheng PP, Kros JM, Li J. Approved CAR T cell therapies: ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov Today. 2018;23:1175–1182. doi:10.1016/j.drudis.2018.02.012.
- Straathof KC, Pulè MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, Heslop HE, Spencer DM, Rooney CM. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105:4247–4254. doi:10.1182/blood-2004-11-4564.
- Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, Yu J, Scholler N, Powell DJ Jr. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012 Apr 1;72(7):1844–1852. Epub 2012 Feb 7. doi:10.1158/0008-5472.CAN-11-3890.
- Kim MS, Ma JS, Yun H, Cao Y, Kim JY, Chi V, Wang D, Woods A, Sherwood L, Caballero D, et al. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J Am Chem Soc. 2015;137:2832–2835. doi:10.1021/jacs.5b00106.
- Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, Schulman A, Du J, Wang F, Singer O, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci U S A. 2016;113:E459–68. doi:10.1073/pnas.1524155113.
- Koristka S, Cartellieri M, Feldmann A, Arndt C, Loff S, Michalk I, Aliperta R, von Bonin M, Bornhäuser M, Ehninger A, et al. Flexible antigen-specific redirection of human regulatory T cells via a novel universal chimeric antigen receptor system. Blood. 2014;124:3494.
- Cartellieri M, Feldmann A, Koristka S, Arndt C, Loff S, Ehninger A, von Bonin M, Bejestani EP, Ehninger G, Bachmann MP. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016;6:e458. doi:10.1038/bcj.2016.61.
- Feldmann A, Arndt C, Bergmann R, Loff S, Cartellieri M, Bachmann D, Aliperta R, Hetzenecker M, Ludwig F, Albert S, et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR”. Oncotarget. 2017;8:31368–31385.
- Albert S, Arndt C, Feldmann A, Bergmann R, Bachmann D, Koristka S, Ludwig F, Ziller-Walter P, Kegler A, Gärtner S, et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. Oncoimmunology. 2017;6:e1287246.
- Mitwasi N, Feldmann A, Bergmann R, Berndt N, Arndt C, Koristka S, Kegler A, Jureczek J, Hoffmann A, Ehninger A, et al. Development of novel target modules for retargeting of UniCAR T cells to GD2 positive tumor cells. Oncotarget. 2017;8:108584–108603.
- Bachmann D, Aliperta R, Bergmann R, Feldmann A, Koristka S, Arndt C, Loff S, Welzel P, Albert S, Kegler A, et al. Retargeting of UniCAR T cells with an in vivo synthesized target module directed against CD19 positive tumor cells. Oncotarget. 2017;9:7487–7500.
- Loureiro LR, Feldmann A, Bergmann R, Koristka S, Berndt N, Arndt C, Pietzsch J, Novo C, Videira P, Bachmann M. Development of a novel target module redirecting UniCAR T cells to Sialyl Tn-expressing tumor cells. Blood Cancer J. 2018;8:81.
- Albert S, Arndt C, Koristka S, Berndt N, Bergmann R, Feldmann A, Schmitz M, Pietzsch J, Steinbach J, Bachmann M. From mono- to bivalent: improving theranostic properties of target modules for redirection of UniCAR T cells against EGFR-expressing tumor cells in vitro and in vivo. Oncotarget. 2018;9:25597–25616.
- Arndt C, Feldmann A, von Bonin M, Cartellieri M, Ewen EM, Koristka S, Michalk I, Stamova S, Berndt N, Gocht A, et al. Costimulation improves the killing capability of T cells redirected to tumor cells expressing low levels of CD33: description of a novel modular targeting system. Leukemia. 2014;28:59–69.
- Koristka S, Cartellieri M, Arndt C, Bippes CC, Feldmann A, Michalk I, Wiefel K, Stamova S, Schmitz M, Ehninger G, et al. Retargeting of regulatory T cells to surface-inducible autoantigen La/SS-B. J Autoimmun. 2013;42:105–116.
- Arndt C, Bachmann M, Bergmann R, Berndt N, Feldmann A, Koristka S. Theranostic CAR T cell Targeting, a brief review. J Labelled Comp Radiopharm. 2019. Epub ahead of print. doi:10.1002/jlcr.3727.
- Arndt C, Feldmann A, Koristka S, Cartellieri M, Dimmel M, Ehninger A, Ehninger G, Bachmann M. Simultaneous targeting of prostate stem cell antigen and prostate-specific membrane antigen improves the killing of prostate cancer cells using a novel modular T cell-retargeting system. Prostate. 2014;74:1335–1346.
- Davenport AJ, Jenkins MR, Ritchie DS, Prince HM, Trapani JA, Kershaw MH, Darcy PK, Neeson PJ. CAR-T cells are serial killers. Oncoimmunology. 2015;4:e1053684.
- Flynn JP, O’Hara MH, Gandhi SJ. Preclinical rationale for combining radiation therapy and immunotherapy beyond checkpoint inhibitors (i.e., CART). Transl Lung Cancer Res. 2017;6:159–168.
- Eckert F, Gaipl US, Niedermann G, Hettich M, Schilbach K, Huber SM, Zips D. Beyond checkpoint inhibition – immunotherapeutical strategies in combination with radiation. Clin Transl Radiat Oncol. 2017;2:29–35.
- Hope TA, Truillet C, Ehman EC, Afshar-Oromieh A, Aggarwal R, Ryan CJ, Carroll PR, Small EJ, Evans MJ. 68Ga-PSMA-11 PET Imaging of Response to Androgen Receptor Inhibition: First Human Experience. J Nucl Med. 2017;58:81–84.
- Feldmann A, Arndt C, Töpfer K, Stamova S, Krone F, Cartellieri M, Koristka S, Michalk I, Lindemann D, Schmitz M, et al. Novel humanized and highly efficient bispecific antibodies mediate killing of prostate stem cell antigen-expressing tumor cells by CD8+ and CD4+ T cells. J Immunol. 2012;189:3249–3259.
- Schäfer M, Bauder-Wüst U, Leotta K, Zoller F, Mier W, Haberkorn U, Eisenhut M, Eder M. A dimerized urea-based inhibitor of the prostate-specific membrane antigen for 68Ga-PET imaging of prostate cancer. EJNMMI Res. 2012;2:23.
- Liolios C, Schäfer M, Haberkorn U, Eder M, Kopka K. Novel Bispecific PSMA/GRPr targeting radioligands with optimized pharmacokinetics for improved PET imaging of prostate cancer. Bioconjug Chem. 2016;27:737–751. Correction: Bioconjug Chem. 2016; 27: 1770.
- Arndt C, von Bonin M, Cartellieri M, Feldmann A, Koristka S, Michalk I, Stamova S, Bornhäuser M, Schmitz M, Ehninger G, et al. Redirection of T cells with a first fully humanized bispecific CD33-CD3 antibody efficiently eliminates AML blasts without harming hematopoietic stem cells. Leukemia. 2013;27:964–967.
- Stamova S, Feldmann A, Cartellieri M, Arndt C, Koristka S, Apel F, Wehner R, Schmitz M, Bornhäuser M, von Bonin M, et al. Generation of single-chain bispecific green fluorescent protein fusion antibodies for imaging of antibody-induced T cell synapses. Anal Biochem. 2012;423:261–268.
- Bippes CC, Feldmann A, Stamova S, Cartellieri M, Schwarzer A, Wehner R, Schmitz M, Rieber EP, Zhao S, Schäkel K, et al. A novel modular antigen delivery system for immuno targeting of human 6-sulfo LacNAc-positive blood dendritic cells (SlanDCs). PLoS One. 2011;6:e16315.
- Carmo-Fonseca M, Pfeifer K, Schröder HC, Vaz MF, Fonseca JE, Müller WE, Bachmann M. Identification of La ribonucleoproteins as a component of interchromatin granules. Exp Cell Res. 1989;185:73–85.
- Feldmann A, Stamova S, Bippes CC, Bartsch H, Wehner R, Schmitz M, Temme A, Cartellieri M, Bachmann M. Retargeting of T cells to prostate stem cell antigen expressing tumor cells: comparison of different antibody formats. Prostate. 2011;71:998–1011.
- Brogsitter C, Zöphel K, Hartmann H, Schottelius M, Wester HJ, Kotzerke J. Twins in spirit part II: DOTATATE and high-affinity DOTATATE–the clinical experience. Eur J Nucl Med Mol Imaging. 2014;41:1158–1165.