
ABSTRACT
An inflammatory microenvironment has been shown to play an important role in the growth and metastasis of tumors. The NLRP3 inflammasome is a multi-protein complex of the innate immune system that is responsible for the production of the potent inflammatory cytokine IL-1β. Tumor- associated macrophages (TAM) are an expanded population of immune cells found in the tumor microenvironment that can promote the initiation and metastasis of tumor cells. Their presence has been correlated with disease burden, highlighting the therapeutic potential of targeting this population. However, to date clinically relevant pharmacologic strategies to target TAM remain elusive. Here, we show that in vitro generated TAM harbor NLRP3 inflammasome components and produce IL-1β. Ibrutinib, an irreversible inhibitor of Bruton’s tyrosine kinase (BTK), is in clinical use for the treatment of B- cell malignancies. We report that BTK is expressed by human in vitro generated TAM and murine macrophages and that it physically associates with the NLRP3 inflammasome. Furthermore, ibrutinib is able to inhibit BTK phosphorylation in TAM generated in vitro. Treatment of TAM with ibrutinib significantly impaired the ability of these cells to produce IL-1β. The present study provides evidence that BTK physically associates with the NLRP3 inflammasome and that inhibition of BTK with ibrutinib can impair the production of IL-1β by in vitro generated TAM. Thus, ibrutinib could potentially be of clinical use in abrogating inflammation-associated cancer progression and the immune-suppressive effects of myeloid cells within the tumor microenvironment.
Introduction
Macrophages are an immune cell population that, while widely distributed among tissues, are highly heterogeneous in phenotype and function. An important function of macrophages is to defend the body against invasive pathogens. However, once they are recruited to the site of a malignant tumor, macrophages can be polarized into tumor-associated macrophages (TAM) by tumor- derived growth factors and function to suppress the host anti-cancer immune response. TAM have been shown to play a functional role in tumor progression via their ability to mediate immune suppression, cancer cell metastasis, and angiogenesis.Citation1,Citation2 This relationship between TAM and the malignant cell makes them an attractive therapeutic target for the treatment of cancer.
The cytokine IL-1β is synthesized as an inactive pro-form that is then processed by a multi-protein sensor of tissue damage and inflammation known as the NLRP3 inflammasome. The NLRP3 inflammasome consists of NLRP3, the adapter protein ASC, and caspase-1 .Citation3 The NLRP3 inflammasome is activated in a two-step process. First, NF-κB signaling is induced through pathogen or danger-associated molecular patterns (PAMP, DAMP) interacting with Toll-like receptor 4 (TLR4), resulting in increased expression of NLRP3 and pro-IL-1β (signal 1). Next, indirect activation of NLRP3 can occur via several different signals (e.g., K+ efflux, Ca2+ signaling, mitochondrial dysfunction, or lysosomal rupture), causing the complex to assemble with and subsequently cleave pro-caspase-1 to give activated caspase-1 (signal 2).Citation4 Activated caspase-1 then cleaves pro-IL-1β to its active form that is then secreted. NLRP3 inflammasome activation was initially shown to be induced by ATP and certain bacterial toxins.Citation5 However, a wide range of alternative stimuli have been identified as activators of the NLRP3 inflammasome, including multiple microbial products, ATP, nigericin, and particulates including alum and uric acid crystals.Citation6–Citation9 The NLRP3 inflammasome has been shown to be a key regulator of the host immune response in the setting of infection, diabetes, and stroke.Citation10,Citation11 Recent work has indicated that the NLRP3 inflammasome may also be a tool by which TAM promote inflammation in the setting of cancer.Citation12,Citation13
Bruton’s tyrosine kinase (BTK) is a Tec family kinase that has long been appreciated for its critical role in B-cell receptor and toll-like receptor signaling.Citation14–Citation20 However, recently our group and others have shown that BTK has a role in modulating myeloid cell signal transduction with important effects on the maturation, trafficking, and function of myeloid cells.Citation21–Citation24 Additionally, BTK inhibition has been shown to impair TNF-α and IL-1β production by monocytes in autoimmune arthritis.Citation25 Given the ability of the NLRP3 inflammasome to synthesize IL-1β, we hypothesized that in vitro generated TAM would express BTK and that BTK would regulate NLRP3 inflammasome function.
Ibrutinib is an irreversible multi-tyrosine kinase inhibitor that covalently binds to a cysteine-481 residue (Cys-481) directly outside of the ATP binding pocket of BTK. This binding event leads to inactivation of the kinase.Citation26 Ibrutinib is currently approved by the FDA for the treatment of several B cell malignancies including chronic lymphocytic leukemia. Previous reports have shown that targeting BTK in malignant B cells inhibits B cell receptor signaling and mediates remarkable regressions of disease.Citation26–Citation28 Therefore, ibrutinib was utilized in this study to effectively inhibit BTK.
In this report, it is demonstrated that human and murine in vitro generated TAM co-express BTK and the NLRP3 inflammasome. Furthermore, ibrutinib treatment inhibited the phosphorylation of BTK and decreased the production of IL-1β by in vitro generated TAM. It was hypothesized that IL-1β production could require the physical interaction of BTK and the NLRP3 inflammasome. Indeed, evidence for co- localization of BTK and the NLRP3 inflammasome in TAM generated in vitro is provided, and this association was inhibited following ibrutinib treatment. Ibrutinib administration also reduced intratumoral levels of IL-1β in vivo. These findings suggest the potential for BTK inhibition to modify the immune profile of cancers via effects on the NLRP3 inflammasome in TAM.
Materials and methods
Cell lines
The MDA-MB 231 human breast adenocarcinoma cell line and the murine macrophage cell line RAW 264.7 were obtained from the American Type Culture Collection. Cells were maintained in DMEM medium containing 10% fetal bovine serum (FBS) and penicillin/streptomycin. The DC9 lymphoblastoid cell line (LCL) was generated by collecting a healthy human donor’s peripheral blood mononuclear cells and infecting the B cells with a strain of Epstein-Barr virus (EBV), B95.8. These B cells were then transformed and became immortalized after the EBV infection and were employed as a lymphoblastoid cell line. These cells were maintained in RPMI 1640 medium containing 10% fetal bovine serum (FBS) and penicillin/streptomycin.
Cytokines
Cytokines employed were recombinant human IL-4 (Shenandoah), recombinant human IL-10 (Peprotech), recombinant human M-CSF (Shenandoah), recombinant human GM-CSF (R&D Systems), recombinant human IL-6 (Shenandoah), and human IFN-γ recombinant protein (ThermoFisher Scientific).
Harvesting tumor-conditioned media
To obtain culture supernatants for the generation of tumor-conditioned media (TCM), approximately 2 × 105 cancer cells per 1 mL were grown to 80% confluence and incubated in 0.2% FBS medium for 24 hours. After incubation, the TCM was harvested and centrifuged to remove suspended cells. The supernatant was collected and 10% FBS was added.
Isolation of human monocytes
Human CD14+ monocytes were prepared from healthy donor blood samples by utilizing the Human RosetteSep Monocyte Enrichment Cocktail CD14+ negative selection (STEMCELL Technologies, Vancouver, BC, Canada) according to the manufacturer’s instruction followed by a Ficoll hypaque density gradient centrifugation technique. Briefly, whole blood was collected into a tube containing heparin and RosetteSep Monocyte Enrichment Cocktail was then added at a concentration of 50 μl/mL of whole blood. After incubation at room temperature for 20 minutes, the blood sample was diluted with an equal volume of PBS and mixed gently. The sample was carefully layered on top of a Ficoll density gradient and centrifuged with the brake off. CD14+ enriched monocytes were removed from the density gradient medium (plasma interface). After washing with PBS, monocyte enriched cells were treated with red blood cell lysis buffer to remove residual red blood cells. The purity of CD14+ monocytes was confirmed via flow cytometric analysis and found to be >94%.
Generation of tumor-associated macrophages
Freshly isolated healthy human CD14+ monocytes were plated in a 100 × 20 mm cell culture dish (USA Scientific) at a concentration of 1 × 106 cells per 1 mL media. To generate TAM, monocytes were cultured in a 1:1 ratio of 10% HAB medium and TCM plus the addition of IL-4 (1 μg/mL), IL- 10 (1 μg/mL), and M-CSF (1 μg/mL). Medium and cytokines were refreshed every other day and cells were harvested on day 7 using a non-enzymatic cell dissociation solution (Sigma Aldrich).Citation2 M1-like macrophages were prepared as described previously.Citation2 In brief, monocytes were cultured in GM-CSF (1 ug/mL) for 7 days. On day 6, media was refreshed with GM-CSF (1 ug/mL), IFN-γ (20 ng/mL), and LPS (50 ng/mL) for 24 hours.
Generation of bone marrow-derived macrophages
Bone marrow was harvested from the femurs of 10 mice and was plated in a 100 × 20 mm cell culture dish (USA Scientific) at a concentration ranging from 1–2 × 106 cells per 1 mL RPMI 1640 media. To generate murine bone marrow-derived macrophages (BMDM), cells were maintained in RPMI 1640 medium containing 10% fetal bovine serum (FBS) and penicillin/streptomycin plus the addition of M-CSF (1 μg/mL). After three days, the cells were replenished with RPMI 1640 medium and M-CSF, and cells were collected on day 7 using a non-enzymatic cell dissociation solution (Sigma Aldrich). The purity of BMDM was confirmed via flow cytometric analysis and found to be between 75–95%.
Immunoblot analysis
Human in vitro generated TAM, RAW 264.7, and DC9 cells were lysed in RIPA buffer (Sigma). Lysates were probed for phosphorylated BTK (p-BTK) (Cell Signaling Technology), total BTK (Cell Signaling Technology), NLRP3 (R&D Systems), ASC (Adipogen), IL-1β (Abcam), or beta-actin (Proteintech) as previously described.Citation29 Where indicated, cells were treated with 0.01% DMSO or 1 μM of ibrutinib (in 0.01% DMSO) for 30 minutes prior to stimulation. To evaluate proteins found in cell supernatants, equal amounts of cells were pretreated with ibrutinib or DMSO for 30 minutes prior to stimulation with LPS (3 hours) and ATP (1 hour). Supernatants were collected and protein was extracted from equal volumes of supernatant. Proteins in the supernatants were precipitated in methanol, chloroform, and diH2O. Precipitated protein was washed in methanol and resuspended in 2X laemmli buffer (BioRad).
Immunofluorescence
Immunofluorescence staining was performed on FFPE tumor tissues via a standard staining procedure using antibodies to the following proteins: IL-1β (Abcam), BTK (Thermofisher), and CD163 (Biorad). TAM generated in vitro were stained with antibodies for phosphorylated p- BTK (R&D Systems), BTK (Thermofisher), NLRP3 (R&D Systems), ASC (Biolegend), IL-6 (Abcam), Alexa Fluor 488 goat anti-mouse and Alexa Fluor 647 goat anti-rabbit (BD Biosciences). Confocal micrographs were taken using the FV1000 Confocal Laser Scanning Microscope (Olympus) with a UPLFLN 60X oil, N.A. 1.3 lens. Tissues or cells were incubated with a non- immune antibody of the same isotype and at the same concentration as the primary antibody, followed by incubation with secondary antibodies. Immunofluorescence staining was quantified using ImageJ software (NIH, Bethesda, Maryland, USA). The quantification of immunofluorescence was expressed as arbitrary unit (AU), which was calculated as (mean pixel intensity X glomerular area)/100,000.
Flow cytometry
Human TAM generated in vitro were treated with DMSO or ibrutinib (1 µM) for 30 minutes followed by stimulation with LPS (50 ng/mL) for 3 hours and ATP (2 mM) for 1 hour. Following stimulation, in vitro generated TAM were permeabilized with eBioscience fixation/permeabilization solution (catalog 00–5523-00) and stained with PE anti-phospho-BTK antibody (Biolegend). Flow cytometry was performed on a LSR II flow cytometer (BD Biosciences, San Jose, CA) and data was analyzed via FlowJo.Citation30
ELISA
Human TAM generated in vitro were treated with DMSO or ibrutinib (1 µM) for 30 minutes followed by stimulation with LPS (50 ng/mL) for 3 hours and ATP (2 mM) for 1 hour. Following stimulation, in vitro generated TAM supernatants were measured for IL-1β by ELISA (R&D Systems) as previously described.Citation30
Real-time PCR
Total RNA was extracted using the TRizol reagent (Life Technologies). Reverse transcription reactions were performed using 500 ng RNA in a 20 μL reaction with the high-capacity reverse transcription kit (Life Technologies). cDNA was used as a template to measure the expression of the human IL-1β gene by quantitative real-time PCR using predesigned primers (Integrated DNA Technologies). Human β-actin served as an internal control for each reaction (Life Technologies). Real-time PCR reactions were performed using the ABI PRISM 7900HT fast Real Time PCR system with SYBR Green chemistry (Applied Biosystems).
Co-immunoprecipitation
Equal amounts of human TAM generated in vitro were treated with DMSO or ibrutinib (1 µM) for 30 minutes followed by stimulation with LPS (50 ng/mL) for 3 hours and ATP (2 mM) for 1 hour. Following stimulation, in vitro generated TAM were lysed in RIPA buffer (Sigma). Cell lysates were incubated with 2 µg of antibody specific for BTK (8547S; Cell Signaling) for 24 hours at 4°C in immunoprecipitation buffer (50 mM Tris [pH 7.3], 150 mM NaCl, 5 mM EDTA, 10 mM NaF, 1% Triton X-100). Cell lysates were then isolated with protein G-Sepharose beads for 2 hours at 4°C. Beads were washed 5 times with cold PBS. Bound proteins were retrieved from G-Sepharose beads by boiling in Laemmli buffer containing β-mercaptoethanol.
In Vivo studies
Female 4–6-week-old BALB/c mice (Jackson Laboratories, Bar Harbor, ME) were purchased and allowed to acclimate for 2–3 weeks before being injected with 106 EMT6 or 105 4T1 cells in the mammary fat pad to produce tumors. Ibrutinib or vehicle was administered in drinking water at 25 mg/kg daily when tumors had reached 100 mm3 in diameter. These studies were conducted under a protocol approved by Ohio State University’s Institutional Animal Care and Use Committee (IACUC 2009A0179-R2).
Immunohistochemistry
Paraffin-embedded tissue was cut at 4 µm and stained with the antibodies to the following proteins: IL-1β (Abcam), NLRP3 (Abcam), and IL-6 (Abcam). Slides were stained using the Intellipath Autostainer Immunostaining System. Tissue and staining were outsourced to our Comparative Pathology and Mouse Phenotyping core that optimized each antibody and utilized the appropriate controls to confirm staining specificity. For quantification, 20X magnification images of tumor tissue (8–10 images per mouse) were captured using PerkinElmerʼs Vectra multispectral slide analysis system. inForm software tools were then used to quantify positive cells within each image as previously described .Citation31
Statistical analysis
Statistical analyzes were performed using GraphPad Prism Software. Statistical significance of differences between groups was analyzed by ANOVA and a two-tailed Student’s t test with P ≤ 0.05 being considered statistically significant.
Ethics approval and consent to participate
The study was initiated after approval from the institutional review board (IRB) and following patient signing of informed consent under an IRB approved protocol for human subject research (IRB protocol 1999C0348).
Results
Human and murine macrophages express bruton’s tyrosine kinase and the NLRP3 inflammasome
The increasing evidence for a role of BTK in myeloid cell differentiation and function led us to hypothesize that TAM would express BTK. To explore this possibility, a differentiation protocol was used to generate a myeloid cell population from peripheral monocytes with characteristics similar to tumor-associated macrophages (TAM). These cells are hereafter referred to as in vitro TAM. BTK expression was measured in TAM generated in vitro,Citation2 murine bone marrow-derived macrophages (BMDM), and the murine macrophage cell line RAW 264.7. BTK was expressed in both human and murine macrophages and all of these cell types expressed BTK at a level similar to those found in control cell line DC9. (–) Given the potential role of BTK as a regulator of the inflammasome, the expression of the inflammasome components NLRP3 and ASC were evaluated in these three cell types. NLRP3 was also expressed in all cell types and ASC was found in TAM generated in vitro and BMDM. However, RAW 264.7 cells lacked high level ASC expressionCitation32 (–). Additionally, human monocytes and in vitro generated M1-like macrophages expressed BTK, NLRP3, and ASC (Figure S1(c–d)). The expression of BTK within tumor-resident macrophages was confirmed in EMT6 murine mammary carcinoma tumors by staining for BTK and a TAM marker (CD163+). Immunofluorescence analysis revealed co- localization of these two markers throughout all sections of tumor that were examined ()). Similar results were obtained when human breast tumors were evaluated for co-localization of BTK and CD163 ()).
Figure 1. Human and murine macrophages express BTK and the NLRP3 inflammasome. (a) Immunoblot for BTK, NLRP3, ASC, and beta-actin (control) expression in TAM generated in vitro. (b) Immunoblot for BTK, NLRP3, ASC, and beta-actin expression in bone marrow-derived macrophages (c) Immunoblot for BTK, NLRP3, ASC, and beta- actin expression in the murine macrophage cell line RAW 264.7. Results are representative of three separate immunoblots in A- C and each lane represents a separate aliquot of cells. (d) Immunofluorescence staining of BTK and CD163 (macrophage marker) in EMT6 murine and human breast tumors. Human breast tumor is from a patient with invasive lobular carcinoma that was positive for estrogen and progesterone receptors and negative for HER 2/neu.
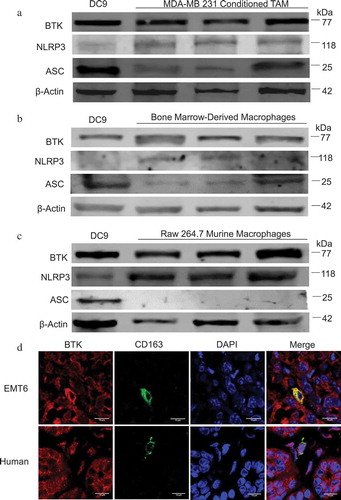
BTK is phosphorylated in TAM generated in vitro following NLRP3 inflammasome activation and its phosphorylation is inhibited by ibrutinib
It was hypothesized that stimulation of the inflammasome would lead to BTK activation and that this effect would be abrogated in the presence of a BTK inhibitor. Therefore, the activation status of BTK in TAM generated in vitro following NLRP3 inflammasome stimulation was evaluated in the presence or absence of ibrutinib, a potent inhibitor of BTK. TAM generated in vitro were primed with LPS for 3 hours (signal 1) followed by stimulation with ATP for 1 hour (signal 2) in order to activate the NLRP3 inflammasome. In vitro generated TAM were pre-treated with either DMSO or ibrutinib (1 µM) prior to inflammasome stimulation and levels of p-BTK were assessed using confocal microscopy, immunoblot analysis, and flow cytometry (–). These experiments revealed that BTK was constitutively phosphorylated in TAM generated in vitro and that levels of p- BTK increased following inflammasome stimulation with LPS and ATP. However, levels of total BTK were not affected (Figure S2(a)). Notably, the activation of BTK was inhibited in the presence of a BTK inhibitor.
Figure 2. BTK is phosphorylated following NLRP3 inflammasome activation and its phosphorylation is inhibited with ibrutinib. (a) Confocal images show in vitro generated TAM stained for p-BTK (green). DAPI nuclear staining appears blue. TAM generated in vitro were pre- treated with either DMSO (control) or ibrutinib (1 µM) for 30 minutes and then stimulated with LPS (50 ng/mL) for 3 hours and ATP (2 mM) for 1 hour. Cells were viewed in 60X and 20X and repeated experimentally three times (b) Immunoblot showing p-BTK and beta-actin (control) expression in TAM generated in vitro and stimulated with LPS and ATP following treatment with either DMSO or ibrutinib (1 µM). Results are representative of three separate immunoblots and quantified in the bar graph. (c) Mean fluorescence intensity of p-BTK (Tyr 223) in TAM generated in vitro treated with DMSO or ibrutinib and stimulated with LPS and then ATP. The results are representative of three separate experiments and quantified in the bar graph. Values are displayed as means ± SE from three independent experiments. Statistical differences between treatment groups were determined using a Student’s t-test, p < .05.
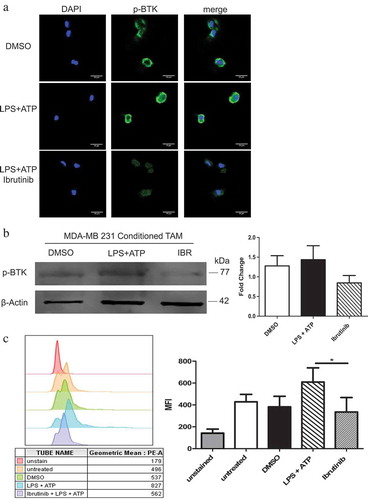
BTK inhibition impairs NLRP3 inflammasome activity
The NLRP3 inflammasome mediates IL-1β production by myeloid cell populations, which could be a potential source of IL-1β within the tumor microenvironment. The ability of BTK inhibition to impair the NLRP3 inflammasome activity in TAM was tested by pre-treating in vitro generated TAM with either DMSO or ibrutinib prior to activation of the NLRP3 inflammasome using LPS and ATP. IL-1β release was measured by ELISA following 4 hours of culture in these conditions. As shown in ), pre-treatment of in vitro generated TAM with a BTK inhibitor led to a significant decrease in IL-1β release compared to control-treated cells. These results were replicated in BMDM (Figure S1(a–b)). A second BTK inhibitor, acalabrutinib,Citation33 had similar inhibitory effects on the release of IL-1β into culture supernatants but was not significant. The effects of BTK inhibition on IL-1β transcript levels was evaluated by qRT-PCR at the 4-hour time point. Despite the observed inhibition of IL-1β production ()), the decreased mRNA levels of IL-1β were not statistically significant. This suggested that BTK inhibition does not markedly affect IL-1β priming ()). The expression of pro- IL-1β and pro-caspase 1 in TAM generated in vitro and RAW 267.4 cells were evaluated under similar conditions (–). Levels of these precursor molecules were not affected by ibrutinib pre-treatments, suggesting that BTK inhibition affects IL- 1β processing rather than priming.Citation11,Citation34 Further evaluation of cleaved IL-1β and caspase-1 in supernatants from TAM generated in vitro confirmed the ability of ibrutinib to impair NLRP3 inflammasome activity ()).
Figure 3. BTK inhibition impairs NLRP3 inflammasome activity. In vitro generated tumor-associated macrophages (TAM) were untreated or treated with either DMSO or ibrutinib (1 μM) for 30 minutes followed by stimulation with LPS (50 ng/mL) for 3 hours and ATP (2 mM) for 1 hour and IL-1β release was analyzed following treatment and stimulation by (a) ELISA and (b) qRT-PCR. Values are displayed as means ± SE from three independent experiments. (c) Immunoblot analysis of pro-IL-1β, pro-caspase-1, and beta-actin (control) expression in TAM generated in vitro (c) and RAW 264.7 cells (d). Cells were treated with either DMSO or ibrutinib (IBR) for 30 minutes followed by stimulation with LPS (50 ng/mL) for 3 hours and ATP (2 mM) for 1 hour. Cells were harvested immediately after ATP. Results are representative of three separate immunoblots and are quantified in the bar graphs as a fold change relative to beta- actin. (e) Cell supernatants from in vitro generated TAM pretreated with either DMSO or ibrutinib followed by stimulation with LPS and ATP were subjected to immunoblot for cleaved IL- 1β and cleaved caspase-1. Results are representative of three separate immunoblots and are quantified in the bar graphs as a fold change relative to DMSO. Statistical differences between treatment groups were determined using a Student’s t-test, * p < .05, ** p < .01, *** p < .001.
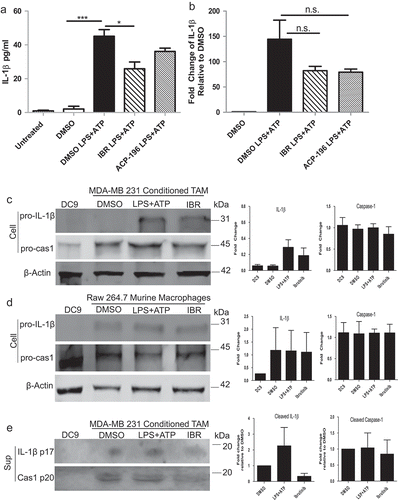
BTK physically interacts with the NLRP3 inflammasome in tumor-associated macrophages generated in vitro and BTK inhibition blocks this event
The ability of ibrutinib to inhibit IL-1β release by in vitro generated TAM led us to hypothesize that BTK plays a critical role in NLRP3 inflammasome function. To test this theory, a co- immunoprecipitation assay was performed in which lysates from activated in vitro generated TAM were processed in order to enrich for BTK and then probed for ASC. As expected, ASC was found to associate with BTK regardless of ibrutinib pre-treatment ()). Furthermore, to confirm the interaction of BTK with the NLRP3 inflammasome, confocal microscopy was utilized to determine co-localization of BTK and NLRP3 inflammasome components NLRP3 and ASC. TAM generated in vitro were pre-treated with DMSO (control) or ibrutinib (1 µM) for 30 minutes followed by stimulation with LPS and ATP. The cells were immunostained with antibodies for BTK (Alexa Fluor 647-red), NLRP3 or ASC (Alexa Fluor 488- green), and DAPI (blue; nuclear staining) and imaged using confocal microscopy. Increased co- localization of BTK and NLRP3 (or ASC) was observed in DMSO treated in vitro generated TAM stimulated with LPS and ATP as compared to in vitro generated TAM pre-treated with ibrutinib suggesting the ability of ibrutinib to block the interaction of the inflammasome and BTK () and Figure S3). The localization of BTK and IL- 6 was evaluated as a control, since this cytokine is not processed by the NLRP3 inflammasome As expected, BTK did not co-localize with IL-6 in LPS/ATP stimulated in vitro generated TAM, nor did BTK inhibition affect the staining results.Citation35
Figure 4. BTK physically interacts with the NLRP3 inflammasome in tumor-associated macrophages generated in vitro. (a) Immunoblots depicting an immunoprecipitation experiment using in vitro generated TAM treated with either DMSO or ibrutinib (IBR) for 30 minutes followed by stimulation with LPS (50 ng/mL) for 3 hours and ATP (2 mM) for 1 hour. Cells were harvested immediately after ATP. Cell lysates were immunoprecipitated for BTK followed by probing for ASC. Results are representative of three separate immunoblots. (b) In vitro generated TAM treated and stimulated as in (a). Cells were stained for BTK (red) and either NLRP3, ASC, or IL-6 control (green) to show co-localization (yellow). DAPI nuclear staining shows as blue. Cells were viewed in 60X and 20X and repeated experimentally three times. Immunofluorescence staining was quantified in the bar graphs and is expressed as arbitrary unit (AU). Values are displayed as means ± SE from 50 cells per group cells. Statistical differences between treatment groups were determined using a Student’s t-test, ***p < .001.
BTK inhibition reduces NLRP3 inflammasome activity in vivo
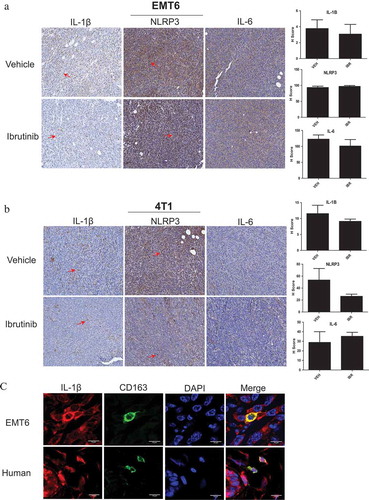
To test the ability of BTK inhibition to reduce NLRP3 inflammasome activity in vivo BALB/c mice inoculated with EMT6 or 4T1 murine mammary carcinoma cells were treated with vehicle or ibrutinib (25 mg/kg daily) via drinking water as previously described.Citation21 At the end of the study, tumors were analyzed for IL-1β, NLRP3, and IL-6 (control) via immunohistochemistry. EMT6 and 4T1 tumors from mice treated with ibrutinib both trended toward decreased IL-1β levels, but neither of these differences were statistically significant. However, no significant change in NLRP3 levels was detected (positive staining is noted by red arrows) (,). The trend in decreased IL-1β in vivo could be explained by a broad number of additional infiltrating cell populations including dendritic and epithelial cells that also express IL-1β and are potentially not affected through BTK inhibition.Citation36–Citation39 However, additional studies are needed to further investigate this hypothesis. Additionally, ibrutinib treated mice also showed a small reduction in tumor volume, but this difference was not statistically significant as we have reported previously.Citation21 As a result, the lack of a difference in tumor volume between vehicle and ibrutinib treated mice suggests that the reduction of IL-1β in vivo is a direct result of ibrutinib therapy, rather than differences in tumor burden. We have also previously shown that neither EMT6 or 4T1 cells express BTKCitation21 which supports the hypothesis that infiltrating macrophages are producing IL- 1β in vivo. To test this, murine EMT6 and human breast tumors were immunostained for CD163 and IL-1β to detect macrophages within the tumor microenvironment ()). Macrophages co- localized with IL-1β staining which suggests that macrophages are a responsible source for IL-1β production in this system.
Figure 5. BTK inhibition reduces NLRP3 inflammasome activity in vivo. Female BALB/c mice were injected with (a) EMT6 or (b) 4T1 cells in the mammary fat pad to produce tumors. n = 8 mice per tumor model. Ibrutinib or vehicle was administered by drinking water at 25 mg/kg daily for 10 days. Tumors were harvested and stained for IL-1β, NLRP3, and IL-6 via immunohistochemistry. Ibrutinib treated mice showed reduced amounts of staining (indicated by red arrow) for IL-1β and NLRP3. IL-6 was used as a control with the expectation that it does not associate with the inflammasome and that its levels should not be affected by BTK inhibition. (c) Immunofluorescence staining of CD163 (marker of macrophages) and IL-1β in EMT6 murine and human breast tumors. Human breast tumor is from a patient with invasive lobular carcinoma that was positive for estrogen and progesterone receptors and negative for HER 2/neu.
Ibrutinib is not cytotoxic and tumor cells do not express BTK
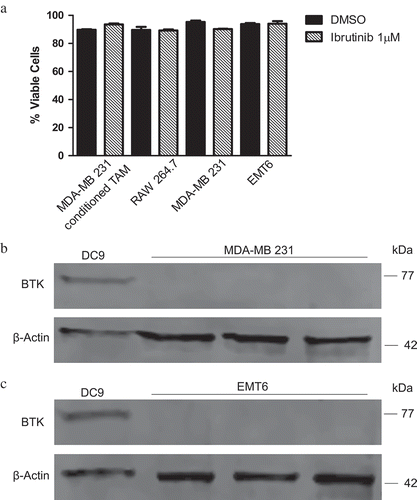
To determine if the effect of ibrutinib on IL-1β production is directly an anti-tumor effect, cell viability was first evaluated. MDA-MB 231-conditioned in vitro generated TAM, RAW 264.7, MDA- MB 231, and EMT6 cells were cultured in either DMSO or ibrutinib (1 μM) for 4 hours, and then viability was assessed using trypan blue staining. Ibrutinib was not cytotoxic to any of these cell types ()). Next, the expression levels of BTK were observed in the breast tumor cell lines MDA-MB 231 (human) and EMT6 (murine). Neither cell line expressed BTK (–). Finally, MDA- MB 231 and EMT6 tumor cells were either untreated or pre-treated with DMSO (control) or ibrutinib (1 µM) for 30 minutes followed by stimulation with LPS and ATP. Following 4 hours of stimulation, cell supernatant was harvested and evaluated for IL-1β production by ELISA. Zero or minimal levels of IL-1β were detected (0–3.4 pg/mL, data not shown). Collectively, these results likely exclude a direct anti-tumor effect of ibrutinib in this model.
Figure 6. Ibrutinib is not cytotoxic. Tumor cells do not express BTK diminishing the likelihood that ibrutinib exerts a direct anti-tumor effect. (a) 5 × 105 MDA- MB 231- conditioned TAM, RAW 264.7, MDA-MB 231, and EMT6 cells were treated with DMSO or ibrutinib (1μM) for 4 hours. After 4 hours cells were mixed with 1-part 0.4% trypan blue to 1-part cell suspension. Viable and nonviable cells were counted. Values represent mean ± SE. This figure is a compilation of three independent experiments. Immunoblot for BTK and beta-actin (control) of (b) MDA-MB 231 and (c) EMT6 cells showing a lack of BTK expression. DC9 cells were used as a positive control.
Discussion
BTK is known to be essential for B cell development and function and has recently been recognized for the role it may play in the function of other immune cell populations.Citation21,Citation22 The present work evaluated the expression of BTK in TAM generated in vitro and the ability of a BTK inhibitor, ibrutinib, to inhibit NLRP3 inflammasome activation and subsequent IL-1β release. BTK was found to be expressed by both human in vitro generated TAM and murine macrophages. Ibrutinib, an FDA-approved BTK inhibitor, was able to inhibit the phosphorylation of BTK in TAM generated in vitro. Furthermore, ibrutinib was able to inhibit the activation of the NLRP3 inflammasome as evidenced by a reduction in IL-1β release by in vitro generated TAM. Ibrutinib appeared to affect the physical interaction of BTK with the NLRP3 inflammasome. Using mouse models of breast cancer, it was shown that ibrutinib treatment resulted in reduced IL-1β expression within the tumor compared to vehicle-treated control mice. Although this decrease was not significant, it could be explained by a broad number of additional infiltrating cell populations including dendritic and epithelial cells that also express IL- 1β and are potentially not affected through BTK inhibition.Citation36–Citation39 However, further studies are needed to further investigate this hypothesis. Although the 4T1 model of breast cancer has substantial levels of neutrophils, this model has been utilized previously to study TAM.Citation36,Citation40–Citation42 Specifically, Makela et al. demonstrated that cell tracking approaches can be utilized to visualize TAM in vivo using the 4T1 model.Citation43 NLRP3 expression within the tumor was not altered by the BTK inhibitor. Of note, IL-1β expression co-localized with macrophages in both human and mouse breast tumors.
The NLRP3 inflammasome is a multi-protein complex that promotes inflammation through activation of caspase-1. The pro-inflammatory cytokine IL-1β is a major product of NLRP3 inflammasome activation.Citation9,Citation44 IL-1β has been hypothesized to be pro-tumorigenic in cancers and has been shown to promote the migration of suppressive myeloid cells including TAM.Citation13,Citation45–Citation47 Increasing evidence supports the concept that inflammation may contribute to the development of human malignancies and alter the host immune response involving macrophages.Citation48–Citation50 Activation of the NLRP3 inflammasome requires two signals. It is hypothesized that in TAM, signal 1 (LPS priming) induces the interaction of BTK with ASC, with signal 2 (ATP) inducing the recruitment of NLRP3 to the BTK-ASC complex, leading to IL-1β and caspase-1 cleavage and release.Citation11 The role of BTK for NLRP3 inflammasome activation has also been explored by Ito et al. who showed BTK as a component of the NLRP3 inflammasome in myeloid cells present at the site of ischemic stroke.Citation11 The use of various deletion mutants of BTK, NLRP3, and ASC enabled them to demonstrate that the ASC-pyrin domain could bind to the BTK-TK domain. Additionally, the NLRP3-NACHT and – leucine-rich repeat domains bound to both the TK and SH2/3 domains of BTK.Citation11
Until recently, BTK was primarily known as a signaling molecule in B cells.Citation51 However, growing evidence supports the role for BTK in myeloid cells including a potential function in Toll-like receptor-mediated recognition of infectious agents, cellular maturation/recruitment processes, and Fc receptor signaling.Citation52 In this study, BTK was also identified as a regulator of innate inflammatory machinery, the NLRP3 inflammasome. In a recent paper by Liu et al., a proteomic screen identified key modulators of the NLRLP3 inflammasome, including BTK.Citation34 Furthermore, Liu et al. found that in vitro administration of ibrutinib to peripheral blood monocular cells from Muckle- Wells syndrome patients showed reduced secretion of IL-1β.Citation34 The current work builds on these studies by showing that in vitro generated TAM express BTK and that ibrutinib can inhibit NLRP3 inflammasome function in TAM generated in vitro. However, this effect may not be exclusively limited to TAM and further studies are needed to determine the effects on other immune cells within the tumor microenvironment.
Our data expands upon what is known about the role of BTK in myeloid cells and highlights BTK as a regulator in TAM pro-inflammatory function. This study has shown the ability of ibrutinib to effectively block the release of IL-1β in human in vitro generated TAM. Targeting the IL-1axis with anakinra (recombinant human IL-1 receptor antagonist IL-1Ra) and canakinumab (neutralizing monoclonal anti-IL-1β antibody) have recently been explored in cancer patients. A therapeutic benefit was achieved with reduced progression of hematological malignancies and also with the improvement of cancer cachexia.Citation53–Citation59 Targeting the NLRP3 inflammasome through BTK may have benefits over other methods of NLRP3 inflammasome inhibition such as MCC950, a NLRP3 inhibitor.Citation60 Although effective, the mechanism and direct target of MCC950 are still unclear. Additionally, many BTK directed therapies are FDA approved in cancer and could be redeployed in the setting of solid tumors.
Given the ability of BTK inhibition to block NLRP 3 inflammasome IL-1β release in TAM generated in vitro, we hypothesize that inhibition of the NLRP3 inflammasome could be used as a therapeutic strategy to target TAM-induced inflammation. In this report, we show that BTK inhibition with ibrutinib can impair NLRP3 inflammasome function. Ibrutinib has the potential to positively impact cancer therapies by targeting TAM and decreasing their activity within the tumor microenvironment.
Conclusion
The present report demonstrates that human and murine TAM generated in vitro express BTK and that ibrutinib is able to inhibit the phosphorylation of BTK in these cells. Treatment of in vitro generated TAM with ibrutinib impaired NLRP3 inflammasome function as evidenced by reduced IL-1β production. Microscopic analysis provided evidence for the existence of a physical interaction between BTK and the inflammasome components NLRP3 and ASC. Importantly, ibrutinib treatment reduced IL-1β expression in both human and murine breast tumor tissue. These results support the investigation of the ability of ibrutinib to influence the immune status of human tumors.
Availability of data and material
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Study Design: BB, LS, AS, MD, LG, JB, ST, WC. Study Conduct: BB, LS, AS, MD, LG, GL. Data Collection: BB, LS, AMH, LG, GL. Data analysis: BB, LS, LG, GL. Manuscript Write Up: All authors. Final Approval of Manuscript: All authors.
Supplemental Material
Download Zip (672 KB)Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Yang L, Zhang Y. Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol. 2017;10:1. doi:10.1186/s13045-017-0430-2.
- Benner B, Scarberry L, Suarez-Kelly LP, Duggan MC, Campbell AR, Smith E, Lapurga G, Jiang K, Butchar JP, Tridandapani S, et al. Generation of monocyte-derived tumor-associated macrophages using tumor-conditioned media provides a novel method to study tumor-associated macrophages in vitro. J ImmunoTher Cancer. 2019;7:140. doi:10.1186/s40425-019-0622-0.
- Lee J, Wan J, Lee L, Peng C, Xie H, Lee C. Study of the NLRP3 inflammasome component genes and downstream cytokines in patients with type 2 diabetes mellitus with carotid atherosclerosis. Lipids Health Dis. 2017;16:217. doi:10.1186/s12944-017-0595-2.
- Ghonime MG, Shamaa OR, Das S, Eldomany RA, Fernandes-Alnemri T, Alnemri ES, Gavrilin MA, Wewers MD. Inflammasome priming by lipopolysaccharide is dependent upon ERK signaling and proteasome function. J Immunol. 2014;192:3881–13. doi:10.4049/jimmunol.1301974.
- Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi:10.1038/nature04515.
- Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem. 1994;269:15195–15203.
- Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol. 2013;191:5230–5238. doi:10.4049/jimmunol.1301490.
- Sha W, Mitoma H, Hanabuchi S, Bao M, Weng L, Sugimoto N, Liu Y, Zhang Z, Zhong J, Sun B, et al. Human NLRP3 inflammasome senses multiple types of bacterial RNAs. Proc Natl Acad Sci U S A. 2014;111:16059–16064. doi:10.1073/pnas.1412487111.
- Broz P, Monack DM. Molecular mechanisms of inflammasome activation during microbial infections. Immunol Rev. 2011;243:174–190. doi:10.1111/j.1600-065X.2011.01041.x.
- Dubois H, Wullaert A, Lamkanfi M. General strategies in inflammasome biology. In: Inflammasome signaling and bacterial infections. Cham: Springer; 2016. p. 1–22. doi: 10.1007/978-3-319-41171-2_1
- Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, Morita R. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun. 2015;6:7360. doi:10.1038/ncomms8360.
- Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73:1907–1916. doi:10.1128/IAI.73.4.1907-1916.2005.
- Elkabets M, Ribeiro VSG, Dinarello CA, Ostrand‐Rosenberg S, Di Santo JP, Apte RN, Vosshenrich CA.IL-1β regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur J Immunol. 2010;40:3347–3357. doi:10.1002/eji.201041037.
- Rip J, de Bruijn MJW, Appelman MK, Pal Singh S, Hendriks RW, Corneth OB.Toll-like receptor signaling drives btk-mediated autoimmune disease. Front Immunol. 2019;10:95. doi:10.3389/fimmu.2019.00095.
- Marron TU. The role of BTK in TLR signaling. J Allergy Clin Immunol. 2009;123:S92–S92. doi:10.1016/j.jaci.2008.12.329.
- Levy O. Bruton tyrosine kinase (Btk): key for signaling via toll-like receptor 8. Blood. 2007;109:2273–2274. doi:10.1182/blood-2006-12-064642.
- Burger JA, Wiestner A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat Rev Cancer. 2018;18:148–167. doi:10.1038/nrc.2017.121.
- Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94:193–205. doi:10.1111/ejh.12427.
- Xia B, Qu F, Yuan T, Zhang Y. Targeting Bruton’s tyrosine kinase signaling as an emerging therapeutic agent of B-cell malignancies. Oncol Lett. 2015;10:3339. doi:10.3892/OL.2015.3802.
- Woyach JA, Johnson AJ, Byrd JC. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood. 2012;120:1175–1184. doi:10.1182/blood-2012-02-362624.
- Stiff A, Trikha P, Wesolowski R, et al. Myeloid-derived suppressor cells express bruton’s tyrosine kinase and can be depleted in tumor-bearing hosts by ibrutinib treatment. Cancer Res. 2016;76:2125–2136. doi:10.1158/0008-5472.CAN-15-1490.
- Fiedler K, Sindrilaru A, Terszowski G, Kokai E, Feyerabend TB, Bullinger L, Rodewald H-R, Brunner C. Neutrophil development and function critically depend on Bruton tyrosine kinase in a mouse model of X-linked agammaglobulinemia. Blood. 2011;117:1329–1339. doi:10.1182/blood-2010-04-281170.
- Mueller H, Stadtmann A, Van Aken H, Hirsch E, Wang D, Ley K, Zarbock A. Tyrosine kinase Btk regulates E-selectin-mediated integrin activation and neutrophil recruitment by controlling phospholipase C (PLC) 2 and PI3K pathways. Blood. 2010;115:3118–3127. doi:10.1182/blood-2009-11-254185.
- Honda F, Kano H, Kanegane H, Nonoyama S, Kim E-S, Lee S-K, Takagi M, Mizutani S, Morio T. The kinase Btk negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils. Nat Immunol. 2012;13:369–378. doi:10.1038/ni.2234.
- Chang BY, Huang MM, Francesco M, Chen J, Sokolove J, Magadala P, Robinson WH, Buggy JJ. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res Ther. 2011;13:R115. doi:10.1186/ar3400.
- Dubovsky JA, Beckwith KA, Natarajan G, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122:2539–2549. doi:10.1182/blood-2013-06-507947.
- Janda E, Palmieri C, Pisano A, Pontoriero M, Iaccino E, Falcone C, Fiume G, Gaspari M, Nevolo M, Di Salle E, et al. Btk regulation in human and mouse B cells via protein kinase C phosphorylation of IBtkγ. Blood. 2011;117:6520–6531. doi:10.1182/blood-2010-09-308080.
- Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298–306. doi:10.1038/nrc3245.
- Duggan MC, Regan-Fendt K, Olaverria GN, Howard JH, Stiff AR, Sabella J, Latchana N, Markowitz J, Gru A, Tridandapani S, et al. Neuroblastoma RAS viral oncogene homolog mRNA is differentially spliced to give five distinct isoforms: implications for melanoma therapy. Melanoma Res. 2019:1. doi:10.1097/CMR.0000000000000623.
- Stiff A, Trikha P, Mundy-Bosse B, McMichael E, Mace TA, Benner B, Kendra K, Campbell A, Gautam S, Abood D, et al. Nitric oxide production by myeloid-derived suppressor cells plays a role in impairing Fc receptor–mediated natural killer cell function. Clin Cancer Res. 2018;24:1891–1904. doi:10.1158/1078-0432.CCR-17-0691.
- Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, Nordquist E, Cruz-Monserrate Z, Yu L, Young G, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut. 2018;67:320–332. doi:10.1136/gutjnl-2016-311585.
- Chalons P, Amor S, Courtaut F, Cantos-Villar E, Richard T, Auger C, Chabert P, Schni-Kerth V, Aires V, Delmas D. Study of potential anti-inflammatory effects of red wine extract and resveratrol through a modulation of interleukin-1-beta in macrophages. Nutrients. 2018:10. doi:10.3390/nu10121856.
- Edlund H, Lee SK, Andrew MA, Slatter JG, Aksenov S, Al-Huniti N. Population pharmacokinetics of the BTK inhibitor acalabrutinib and its active metabolite in healthy volunteers and patients with B-cell malignancies. Clin Pharmacokinet. 2018. doi:10.1007/s40262-018-0725-7.
- Liu X, Pichulik T, Wolz -O-O, Dang T-M, Stutz A, Dillen C, Delmiro Garcia M, Kraus H, Dickhöfer S, Daiber E, et al. Human NACHT, LRR, and PYD domain–containing protein 3 (NLRP3) inflammasome activity is regulated by and potentially targetable through Bruton tyrosine kinase. J Allergy Clin Immunol. 2017;140:1054–1067.e10. doi:10.1016/j.jaci.2017.01.017.
- Horwood NJ, Page TH, McDaid JP, Palmer CD, Campbell J, Mahon T, Brennan FM, Webster D, Foxwell BMJ. Bruton’s tyrosine kinase is required for TLR2 and TLR4-induced TNF, but not IL-6, production. J Immunol. 2006;176:3635–3641. doi:10.4049/jimmunol.176.6.3635.
- Kakoschky B, Pleli T, Schmithals C, Zeuzem S, Brüne B, Vogl TJ, Korf H-W, Weigert A, Piiper A, Mondelli MU. Selective targeting of tumor associated macrophages in different tumor models. PLoS One. 2018;13:e0193015. doi:10.1371/journal.pone.0193015.
- Eriksson U, Kurrer MO, Sonderegger I, et al. Activation of dendritic cells through the interleukin 1 receptor 1 is critical for the induction of autoimmune myocarditis. J Exp Med. 2003;197:323–331. doi:10.1084/jem.20021788.
- Bent R, Moll L, Grabbe S, Bros M. Interleukin-1 beta-A friend or foe in malignancies? Int J Mol Sci. 2018:19. doi:10.3390/ijms19082155.
- Guo B, Fu S, Zhang J, Liu B, Li Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci Rep. 2016;6:36107. doi:10.1038/srep36107.
- Georgoudaki A-M, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Östling J, Dahan R, Harris R, Rantalainen M, Klevebring D, et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 2016;15:2000–2011. doi:10.1016/j.celrep.2016.04.084.
- Luo Y, Zhou H, Krueger J, et al. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J Clin Invest. 2006;116:2132–2141. doi:10.1172/JCI27648.
- Duan X, He K, Li J, Cheng M, Song H, Liu J, Liu P. Tumor associated macrophages deliver iron to tumor cells via Lcn2. Int J Physiol Pathophysiol Pharmacol. 2018;10:105.
- Makela AV, Gaudet JM, Foster PJ. Quantifying tumor associated macrophages in breast cancer: a comparison of iron and fluorine-based MRI cell tracking. Sci Rep. 2017;7:42109. doi:10.1038/srep42109.
- Moossavi M, Parsamanesh N, Bahrami A, Atkin SL, Sahebkar A. Role of the NLRP3 inflammasome in cancer. Mol Cancer. 2018;17:158. doi:10.1186/s12943-018-0900-3.
- Kolb R, Liu G-H, Janowski AM, Sutterwala FS, Zhang W. Inflammasomes in cancer: a double-edged sword. Protein Cell. 2014;5:12–20. doi:10.1007/s13238-013-0001-4.
- Umansky V, Sevko A. Tumor microenvironment and myeloid-derived suppressor cells. Cancer Microenviron. 2013;6:169–177. doi:10.1007/s12307-012-0126-7.
- Najjar YG, Rayman P, Jia X, Pavicic PG, Rini BI, Tannenbaum C, Ko J, Haywood S, Cohen P, Hamilton T, et al. Myeloid-derived suppressor cell subset accumulation in renal cell carcinoma parenchyma is associated with intratumoral expression of IL1β, IL8, CXCL5, and Mip-1α. Clin Cancer Res. 2017;23:2346–2355. doi:10.1158/1078-0432.CCR-15-1823.
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi:10.1016/j.cell.2010.01.025.
- Perwez Hussain S, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007;121:2373–2380. doi:10.1002/ijc.23173.
- Berraondo P, Minute L, Ajona D, Corrales L, Melero I, Pio R. Innate immune mediators in cancer: between defense and resistance. Immunol Rev. 2016;274:290–306. doi:10.1111/imr.12464.
- Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol Cancer. 2018;17:57. doi:10.1186/s12943-018-0779-z.
- Weber ANR, Bittner Z, Liu X, Dang T-M, Radsak MP, Brunner C. Bruton’s tyrosine kinase: an emerging key player in innate immunity. Front Immunol. 2017;8:1454. doi:10.3389/fimmu.2017.01454.
- Lust JA, Lacy MQ, Zeldenrust SR, Witzig TE, Moon-Tasson LL, Dinarello CA, Donovan KA. Reduction in C-reactive protein indicates successful targeting of the IL-1/IL-6 axis resulting in improved survival in early stage multiple myeloma. Am J Hematol. 2016;91:571–574. doi:10.1002/ajh.24352.
- Lust JA, Lacy MQ, Zeldenrust SR, Dispenzieri A, Gertz MA, Witzig TE, Kumar S, Hayman SR, Russell SJ, Buadi FK, et al. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1β-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin Proc. 2009;84:114–122. doi:10.4065/84.2.114.
- Wu Z, Han X, Qin S, Zheng Q, Wang Z, Xiang D, Zhang J, Lu H, Wu M, Zhu S, et al. Interleukin 1 receptor antagonist reduces lethality and intestinal toxicity of 5-fluorouracil in a mouse mucositis model. Biomed Pharmacother. 2010;64:589–593. doi:10.1016/j.biopha.2010.06.006.
- Wang X, Gao J, Qian L, Gao J, Zhu S, Wu M, Zhang Y, Guan W, Ye H, Yu Y, et al. Exogenous IL-1Ra attenuates intestinal mucositis induced by oxaliplatin and 5-fluorouracil through suppression of p53-dependent apoptosis. Anticancer Drugs. 2015;26:35–45. doi:10.1097/CAD.0000000000000142.
- Sauter KAD, Wood LJ, Wong J, Iordanov M, Magun BE. Doxorubicin and daunorubicin induce processing and release of interleukin-1β through activation of the NLRP3 inflammasome. Cancer Biol Ther. 2011;11:1008–1016. doi:10.4161/cbt.11.12.15540.
- Hickish T, Andre T, Wyrwicz L, et al. MABp1 as a novel antibody treatment for advanced colorectal cancer: a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2017;18:192–201. doi:10.1016/S1470-2045(17)30006-2.
- De Mooij CEM, Netea MG, Van Der Velden WJFM, Blijlevens NMA. Targeting the interleukin-1 pathway in patients with hematological disorders. Blood. 2017. doi:10.1182/blood-2016-12-754994.
- Perera AP, Fernando R, Shinde T, Gundamaraju R, Southam B, Sohal SS, Robertson AAB, Schroder K, Kunde D, Eri R. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci Rep. 2018;8:8618. doi:10.1038/s41598-018-26775-w.