
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Targeted immunotherapy of solid cancers with chimeric antigen receptor (CAR) T cells and immunocytokines are attractive options in that they both rely on the specificity of tumor-targeted antibodies. Since carcinoembryonic antigen (CEA) expression in both colon and breast cancers is correlated with poor prognosis, it was chosen as a model tumor target in immunocompetent CEA transgenic (CEATg) mice. A second-generation anti-CEA CAR derived from CEA-specific antibody T84.66 was used to treat murine MC38 colon or E0771 breast carcinomas transfected with CEA. Anti-CEA CAR vs. mock transduced T cells exhibited a CEA-specific cytotoxic and IFN dose response to both CEA transfected cell lines vs. their CEA-negative controls. Anti-CEA CAR vs. mock transduced T cells delayed the median survival of CEA transfected s.c. MC38 or orthotopic E0771 tumor-bearing CEATg mice by 2 days. With the addition of one-day prior cyclophosphamide (CY) lymphodepletion, anti-CEA CAR T cell treatment delayed the median survival of MC38/CEA and E0771/CEA tumor-bearing CEATg mice by ten and 3 days, respectively. Since CAR T cells require IL2 for survival and expansion, anti-CEA-IL2 immunocytokine (ICK) treatment was performed post CAR T cell therapy. Single ICK treatment 1 day after CY plus anti-CEA CAR T cell therapy in the MC38/CEA model, and two ICK treatments every 3 days after CY plus anti-CEA CAR T cell therapy in the E0771/CEA model were ineffective, while four ICK treatments every 3 days after CY plus anti-CEA CAR T cell therapy completely eradicated MC38/CEA tumor growth and induced tumor immunity when the mice were re-challenged with tumor. These studies show the therapeutic potential of anti-CEA CAR T cells combined with ICK to treat CEA-positive tumors.
Abbreviations: CAR: Chimeric antigen receptor, CEA: Carcinoembryonic antigen, CEACAM5, ICK: Immunocytokine, CY: Cyclophosphamide, CEATg mouse: transgenic CEA mouse, TDLN: Tumor-draining lymph node
Introduction
Colon cancer is the third leading cause of cancer-related deaths in men and women,Citation1,Citation2 and breast cancer is the second most common cancer for women in the United States.Citation3 The advent of targeted immunotherapy has played a major role in new approaches to the treatment of these cancers while reducing side effects associated with untargeted therapies.Citation4–6 In terms of antibody-based tumor targeting, a potential target for both colon and breast cancers is carcinoembryonic antigen (CEA). CEA is expressed in over 90% of colonCitation7 and about 50% of breastCitation8–11 cancers; and high serum CEA levels have been correlated with poor prognosis in both colon and breast cancer patients.Citation12–22 Although CEA is expressed in normal colon, CEA expression is polarized to the apical side of epithelial cells and is not accessible to circulating antibodies;Citation23 however, during tumor invasion, CEA expression on tumor cells becomes accessible to systemic antibodies as evidenced by targeting with radiolabeled anti-CEA antibodies.Citation24–26
Tumor antigens such as CEA that are expressed in normal colon are poorly immunogenic but can be retargeted by the use of chimeric antigen receptors (CARs) expressed on T cells since they are derived from monoclonal antibodies that were produced by immunization of mice with a human antigen.Citation27–30 Similarly, endogenous T cells do not target CEA, but CAR T cells break tolerance by using CARs derived from monoclonal antibodies and bypass the requirement for antigen presentation in the context of MHC.Citation31 In a pre-clinical study, Chmielewski et al. showed that anti-CEA CAR T cells reduced the size of orthotopic CEA-positive pancreatic tumors in immunocompetent CEA transgenic (CEATg) mice without inducing tissue damage to CEA-positive organs such as the normal colon, despite the infiltration of anti-CEA CAR T cells.Citation32 In clinical studies, Katz et al. showed that anti-CEA CAR T cell therapy did not induce therapy-related grade 4 or 5 adverse events in patients with CEA-positive liver metastases.Citation33 Nonetheless, CAR T therapy alone for solid tumors is relatively ineffective and requires additional support. In an attempt to improve tumor responses, combination therapy of anti-CEA CAR T cell therapy plus systemic IL2 was attempted but had IL2-related toxicity preventing further treatment. The issue of systemic IL2 toxicityCitation33 can be reduced by genetic fusion of tumor antigen-specific antibodies to form immunocytokines (ICKs). We have previously shown that the ICK, humanized anti-CEA antibody (M5A) fused to IL2, was effective against orthotopic CEA transfected breast carcinoma E0771 tumors in a CEATg mouse model.Citation34 Furthermore, combination of ICK with fractionated stereotactic radiation therapy (SRT) eradicated tumors and established tumor immunity. These studies prompted us to combine two antigen-targeted therapies, namely anti-CEA CAR T cells plus ICK therapy to determine their therapeutic efficacy.
Materials and methods
DNA constructs
Retrovirus vector expressing GFP tag (pMIGR) was supplied as a gift from Dr. Zuoming Sun (City of Hope).Citation35 The GFP tag was removed from pMIGR via restriction enzyme digestion at NcoI and PacI and ligation with a CCTGAA insert between the two restriction enzyme sites (pMSCV). The scFv sequence used in the CAR construct was obtained from the murine anti-CEA T84.66 monoclonal antibody.Citation36 The second-generation CAR construct consisted of the T84.66 scFv (VL-(GGGGS)3-VH) fused to a 129-amino acid middle-length CH2-deleted version of the IgG4 Fc spacerCitation37 with the intracellular co-stimulatory signaling murine CD28 transmembrane and murine CD3ζ cytolytic domains. As a transduction marker, the ectodomain of mouse CD19 (mCD19t), was added downstream of the same promoter as anti-CEA CAR.Citation37–39 T2ACitation40 was also inserted between anti-CEA CAR and mCD19t. The anti-CEA CAR construct was inserted into pMSCV vector via restriction enzyme digestion and ligation (pMSCV_anti-CEA CAR_mCD19t). As a control, mCD19t was inserted into pMSCV via PCR, restriction enzyme digestion, and ligation (pMSCV_mCD19t). After ligation, the insert was sequenced to ensure that there were no PCR-induced mutations. The constructs are shown in (). The immunocytokine (ICK) was produced as described by Kujawski et al.Citation34
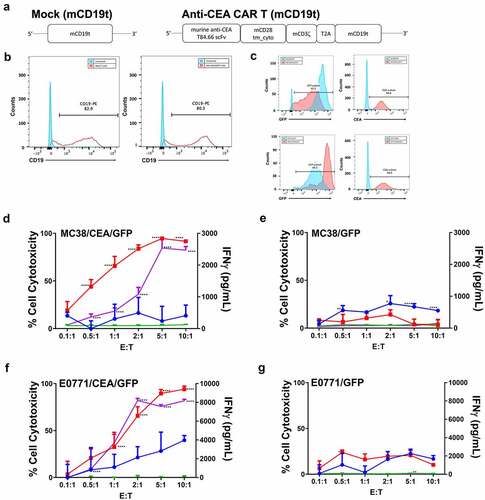
Figure 1. Anti-CEA CAR T cells specifically target CEA-positive mouse adenocarcinoma cells in vitro.
Cell lines and culture conditions
The Platinum-E retroviral packaging (PlatE) cell line (Cell Biolabs, RV-101) was grown in Dulbecco’s Modified Eagle Medium (DMEM; Corning, 10–013-CV) supplemented with 10% fetal bovine serum (FBS), antibiotic-antimycotic solution (Corning, MT30004), 1 μg/mL puromycin (InvivoGen, ant-pr-1), and 10 μg/mL blasticidin (InvivoGen, ant-bl-1). Mouse colon adenocarcinoma cell lines with or without CEA expression (MC38/CEA or MC38, respectively) were grown in DMEM supplemented with 10% FBS, and antibiotic-antimycotic solution. GFP-expressing mouse colon adenocarcinoma cell line (MC38/GFP) and GFP-expressing mouse breast adenocarcinoma cell line (E0771/GFP) were grown in DMEM supplemented with 10% FBS, antibiotic-antimycotic solution, and 1 μg/mL puromycin. GFP-expressing, CEA-positive mouse colon adenocarcinoma cell line (MC38/CEA/GFP) and GFP-expressing, CEA-positive mouse breast adenocarcinoma cell line (E0771/CEA/GFP) were grown in DMEM supplemented with 10% FBS, antibiotic-antimycotic solution, and G418 Sulfate.
Virus production/transfection
PlatE cells at 80–90% confluency were transfected with either pMSCV_mCD19t or pMSCV_anti-CEA CAR_mCD19t plasmid. Prior to transfection, the medium for each plate was changed from DMEM supplemented with 10% FBS, antibiotic-antimycotic solution, 1 μg/mL puromycin, and 10 μg/mL blasticidin to DMEM supplemented with 10% FBS. Then, each plate was transfected with 14 μg of total DNA, 20 μL of PLUS solution, and 20 μL of lipofectamine LTX (Invitrogen, 15338–100) in 200 μL serum reduced OPTI-MEM (Gibco, 31985070), as per manufacturer’s directions. After 24 hours from the transfection, the medium was aspirated and replaced with new DMEM supplemented with 10% FBS. After 48 and 72 hours from the transfection, the supernatants were collected with 30 mL syringe (BD, 302833) and filtered through 0.45 μm syringe filter (Millipore Sigma, SLHV033RS). Retrovirus in the collected supernatant was either used immediately or frozen at −80°C for later use.
Mouse primary CD3+ T cells isolation and activation
Spleens from CEATg mice were separated into single-cell suspension using 40 μm cell strainers (Falcon, 352340). The red blood cells were lysed using Red Blood Cell Lysis Buffer Hybri-Max (Sigma, R7757), per manufacturer’s directions. The CD3+ cells were isolated from the primary splenocytes using EasySep Mouse T Cell Isolation Kit (StemCell, 19851), per manufacturer’s directions. The CD3+ cells were mixed with Dynabeads Mouse T-Activator CD3/CD28 for T cell Expansion and Activation (Thermo Fisher Scientific, 11453D), per manufacturer’s directions. Activation of CD3+ cells was performed in RPMI 1640 (Gibco, 21870076) with 10% FBS, 2 mM L-glutamine, antibiotic-antimycotic solution, 10 ng/mL recombinant mouse IL7 (R&D, 407-ML), 50 IU/mL recombinant mouse IL2 (Biolegends, 575402), and 5.5 mM β-mercaptoethanol (Gibco, 21985–023) and plated at 1 × 106 cells/well in 24 wells plate (Corning, T-2989-24) overnight at 37°C.
Transduction of activated mouse primary CD3+ T cells
On Day 0, 24 well plates pre-coated with 20 µg/well retronectin (Takara, T100B) were incubated overnight at 4°C, per manufacturer’s directions. On Day 1, the activated CD3+ cells with Dynabeads Mouse T-Activator CD3/CD28 were infected in retronectin pre-coated 24 wells plate with retrovirus containing mCD19t (mock T cells) or anti-CEA CAR. Recombinant mouse IL7 (10 ng/mL), recombinant mouse IL2 (50 IU/mL), and β-mercaptoethanol (5.5 mM) were added to retrovirus before transducing CD3+ cells. The MOI was 0.5. The transduced CD3+ cells were incubated overnight at 37°C. On Day 2, retrovirus was removed from each well and replaced with fresh culture media of RPMI 1640 with 10% FBS, 2 mM L-glutamine, antibiotic-antimycotic solution, recombinant mouse IL7, recombinant mouse IL2, and β-mercaptoethanol and plated at 0.5 × 106 cells/well in 24 wells plate overnight. On Day 4, transduced CD3+ cells were collected. The Dynabeads Mouse T-Activator CD3/CD28 were removed from collected transduced CD3+ cells (StemCell, 18000). Truncated mouse CD19-positive CD3+ cells were positively selected using mouse CD19 Positive Selection Kit II (StemCell, 18954), per manufacturer’s directions, and plated at 0.5 × 106 cells/well in 24 wells plate overnight. On Day 5, truncated mouse CD19-positive CD3+ cells were collected and resuspended into 1 × 106 cells/mL concentration for in vitro experiments and 2.5 × 107 cells/mL concentration for in vivo experiments.
In vitro killing assay
Mock or anti-CEA CAR T cells were incubated with either MC38/GFP, MC38/CEA/GFP, E0771/GFP, or E0771/CEA/GFP cells at 0.1:1, 0.5:1, 1:1, 2:1, 5:1, or 10:1 ratio (Effectors:Targets, E:T) in RPMI 1640 medium, no phenol red (Gibco, 11835030) with 10% FBS, 2 mM L-glutamine, antibiotic-antimycotic solution, and β-mercaptoethanol on 96 wells (Eppendorf, 951040145) overnight at 37°C. For controls, only target cells were plated. For immunocytokine (ICK) studies, ICK (12 ng/mL; equivalent to 1 ng/mL IL2) was added to media along with T cells and target cells at the beginning of 24 hours co-culture. For positive controls, 20% Triton x-100 (Sigma-Aldrich, X100-100ML) in PBS was added to wells with only target cells and incubated for 30 minutes at 37°C. Supernatants were removed and collected for measurement of IFNγ by ELISA. For quantitative analysis of GFP, culture media were replaced with fresh 200 µL of RPMI 1640 medium without phenol red and GFP fluorescence was read on a CLAROstar instrument. Mouse IFNγ ELISA (BioLegends, 430806) was performed per manufacturer’s directions. Supernatants were collected from each well of in vitro killing assay plate and diluted 1:5 for IFNγ ELISA.
Cytotoxic activity
To calculate the percentage of cell cytotoxicity, the value for 100% cell viability (valuemax) was measured by averaging the fluorescence readings of target cells cultured without any T cells. The fluorescence measured for each well co-cultured with effector and target cells is the experimental value (valueexp). The background was subtracted as follows, valuemin from both valuemax and valueexp. The minimum value (valuemin) was calculated by taking the average of fluorescence measured for target cells cultured without T cells and lysed by trypsin-EDTA. The fraction of live cell fluorescence was calculated by dividing (valueexp – valuemin) by (valuemax – valuemin). Then, the fraction of live cell fluorescence was subtracted from 1 to correspond to the fluorescence lost by GFP-expressing target cell death. The percentage of cell cytotoxicity in each well was calculated by multiplying by 100%.
Flow cytometry
For flow cytometry, cells were resuspended in PBS-2%FBS solution. Cells were incubated with appropriate staining antibody conjugated with fluorophore for 30 minutes at 4°C in dark. T cells were stained with FITC anti-mouse CD8a (BD Biosciences, 553031), PE/Cy7 anti-mouse CD4 (Biolegend, 100422), PE anti-mouse IFNγ (Biolegend, 505808), APC anti-mouse PD-1 (Biolegend, 135210), Brilliant Violet 421 anti-mouse CD127 (Biolegend, 135024), FITC anti-mouse CD19 (Biolegend, 152404), PE anti-mouse CD19 (Biolegend, 152408), PerCP/Cy5.5 anti-mouse CD4 (Biolegend, 100434), PE anti-mouse CD8b.2 (Biolegend, 140408), APC Rat Anti-Mouse CD4 (BD, 561091), FITC Rat Anti-Mouse CD8a (BD, 553030), Horizon BV711 Rat Anti-Mouse CD19 (BD, 563157), PE Rat Anti-Mouse CD4 (BD, 553049), PE-Cy7 Rat Anti-Mouse CD25 (BD, 552880), or APC Hamster Anti-Mouse CD279 (BD, 562671). T cells were stained with Anti-Mouse/Rat FoxP3 Staining Set PE (eBioscience, 72–5775-40), per manufacturer’s directions. Mouse adenocarcinoma cells were stained with human CEACAM-5/CD66e APC-conjugated antibody (R&D, FAB412181A). Mouse cells were stained with Pacific Blue Annexin V (Biolegend, 640918) or Horizon Fixable Viability Stain 510 (BD, 564406) for cell viability. Flow cytometry was performed on LSRFortessa (BD) and analyzed by FlowJo software (v9 and v10). The multi-color panel was compensated with Anti-Rat and Anti-Hamster Igκ/Negative Control Compensation Particles Set (BD, 552845) and Anti-Mouse Ig, κ/Negative Control Compensation Particles Set (BD, 552843).
Animal model, tumor challenge, and treatment
Mouse care and experimental procedures were performed under pathogen-free conditions in accordance with the Institutional Animal Care and Use Committee of City of Hope (IACUC protocol number 16103). The CEA transgenic mice were previous generated by inserting a 32.6-kb fragment containing the complete human CEA gene and flanking sequences isolated from a genomic cosmid clone and used to produce transgenic C57BL/6 mice as previously described.Citation41 A homozygous line was established that was designated C57BL/6 J-TgN(CEAGe)18FJP. Southern blot analysis showed that this line contained intact copies of the cosmid clone, with approximately 19 integrated copies at one chromosomal location. A mouse-human chimeric anti-CEA monoclonal antibody was used to examine CEA expression by immunohistochemical staining of frozen tissue sections. In the cecum and colon, approximately 20% of the luminal epithelial cells had strong cytoplasmic staining, whereas occasional glands showed intense staining. CEA was also expressed in gastric foveolar cells, whereas small intestine villi had only a few (<1%) positive cells. CEA was not found by immunohistochemistry in other tissues of the digestive tract, nor was it found in a wide range of other tissues or organs.
On Day 1, 1 × 106 MC38/CEA or 1 × 105 E0771/CEA cancer cells in 50–100 μL of PBS combined with 25 μL of Matrigel (Corning, 356237) were injected s.c. or orthotopically into CEATg mouse using 28 G Insulin Syringes (BD, 329461). Tumor size was measured along with each mouse’s weight. Established tumors (50–75 mm3) were treated with 5 × 106 Mock or anti-CEA CAR T cells in 200 μL PBS were injected i.v. into CEATg mice. For multiple anti-CEA CAR T cell therapy group, a second injection was performed 7 days after the first injection with third injection was given 14 days later. Depending on the study groups, lymphodepletion was induced by i.p. injection of 100 mg/kg cyclophosphamide monohydrate (Sigma, C7397-1 G) 1 day before the start of T cell therapy. For single ICK treatment groups, 25 μg ICK was i.p. injected 1 day after T cell therapy. For the multiple ICK treatment groups, 25 μg ICK was i.p. injected 1 day after T cells and repeated every 3 days.
Tissue collection and analysis
Survival studies were terminated for tumors >1500 mm3. Colon, small intestine, large intestine, stomach, liver, kidneys, spleen, lung, and heart were collected in cold PBS. Blood was collected in 0.5 M EDTA (Invitrogen 15575020). Tissues were washed in PBS and frozen on dry ice using O.C.T. (FisherHealthCare, 4585) in vinyl Specimen molds (Sakura, 4557) for H&E and IHC staining. For flow cytometry analysis, small fractions of spleen were cut and meshed on 0.40 μm cell strainer (Corning, 431750) before lysing in Red Blood Cell Lysis Buffer Hybri-Max (Sigma, R7757). Blood was lysed in Red Blood Cell Lysis Buffer Hybri-Max and resuspended in PBS-2%FBS. Tumor was cut in small pieces and digested with Tumor Dissociation Kit, Mouse (MACS, 130–096-730) and gentleMACS C Tubes (MACS, 130–096-334), as per manufacturer’s directions. The cells were stained with fluorescent antibodies for flow cytometry. After the surface proteins were stained, intercellular expression of FoxP3 was stained with Anti-Mouse/Rat Foxp3 Staining Set PE (eBioscience, 72–5775-40), as per manufacturer’s directions. For IFNγ production cells were re-stimulated using PMA (10 ng/ml; Sigma-Aldrich) and ionomycin (1 µg/ml; Sigma-Aldrich) in the presence of Brefeldin A (5 µg/ml; BioLegend, CA) in 10% FBS IMDM media for 4 hours in 37°C. Next, cells were stained for surface markers and viability marker (Zombie UV, BioLegend) and fixed and permeabilized using Foxp3 Transcription Factor Fixation/Permeabilization kit (ThermoFisher) following the manufacture’s protocol and stained for intracellular IFNγ (BioLegend) and analyzed by flow cytometry.
Statistical analysis
Cell cytotoxicity, cytokine, and mean tumor volume measurements, as well as survival over time, were analyzed via Prism software using the T-test, one-way ANOVA, or two-way ANOVA analysis.
Results
Target specificity of anti-CEA CAR T cells
An all murine anti-CEA second-generation CAR consisting of a single-chain variable fragment (scFv) derived from the anti-CEA T84.66 monoclonal antibodyCitation42 fused to a CH2 domain deleted IgG4 Fc murine CD28 transmembrane and CD3ζ was constructed to allow detection of anti-CEA CAR expression on mouse T cell surface (). CD3+ T cells were transduced with mCD19t (mock T cells) or anti-CEA CAR with mCD19t (anti-CEA CAR T cells) and expression confirmed by detection of CD19 ().
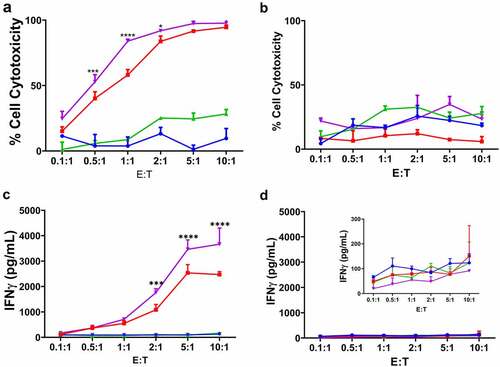
To determine the antigen-specificity of anti-CEA CAR T cells, anti-CEA CAR or mock transduced T cells were incubated with murine MC38 colon and E0771 breast adenocarcinoma cell lines transfected with CEA and GFP in increasing Effector:Target (E:T) ratios with GFP only as a positive control. GFP and CEA expression were confirmed by flow cytometric detection (). Specific lysis of CEA+ vs. CEA− target cells with anti-CEA CAR T cells was demonstrated, while mock transduced T cells were ineffective in killing targets (-g). For both CEA+ target cells, only anti-CEA CAR T cells showed a dose response of increasing levels of IFNγ with increasing E:T ratios (). Together, these data confirmed that anti-CEA CAR T cells specifically target CEA-expressing cells via antigen recognition and T cell activation.
Delay of CEA+ colon adenocarcinoma tumor growth by anti-CEA CAR T therapy
To evaluate the therapeutic potential of anti-CEA CAR T cells, MC38/CEA colon tumors were implanted in immunocompetent CEA transgenic (CEATg) mice and treated with a single i.v. injection of either anti-CEA CAR or mock transduced T cells. There was a significant reduction in tumor growth in anti-CEA CAR T cell treated mice compared to mock transduced T cell treated mice up to day 24 (Supplement ). After Day 24, a number of mice treated with mock transduced T cells had to be euthanized due to large tumor size. One out of 5 mice treated with anti-CEA CAR T cells exhibited tumor regression out to day 54 with subsequent tumor regrowth followed out to day 71 post tumor implantation (Supplement ). The median overall survival for mice treated with anti-CEA CAR T cells was statistically significant compared to mice treated with Mock T cells (p < .02, Supplement ). No whole-body toxicity was observed as measured by mouse weight (Supplement ), as well as absence of diarrhea or loss of physical mobility. These data indicate that systemic delivery of anti-CEA CAR T cells in this model resulted in modest delays in CEA+ tumor growth without severe toxicity in immune-competent CEATg mice. This is in agreement with other studies that have shown that CAR T cell therapy alone was not sufficient to eradicate solid tumors.Citation43
Immunodepletion improves the efficacy of anti-CEA CAR T cell therapy
Since the administration of exogenous T cells in immunocompetent mice leads to homeostatic reduction of T cells,Citation44–46 T cell depletion by short-acting agents such as cyclophosphoamide (CY) can improve CAR T therapy.Citation47–49 CY, an alkylating agent, is a commonly used chemotherapeutic agentCitation50 that selectively depletes immunosuppressive cells, such as regulatory T cells, to increase antitumor activity.Citation48,Citation51 While CY treatment alone is also not sufficient to eradicate most solid tumors, it was quite effective in the treatment of MC38 tumors as shown by Myers et. al.Citation52 Although MC38/CEA tumors were treated with the same doses of CY as MC38 tumors, MC38/CEA tumors were resistant to CY treatment (Supplement and Supplement ), in agreement with the chemoresistant effect of CEA expression in colon cancers.Citation53 For this reason the combination therapy was performed only on mice bearing MC38/CEA tumors.
Figure 2. Improved effect of cyclophosphamide plus anti-CEA CAR T cells on s.c. MC38/CEA and orthotopic E0771/CEA tumor growth inhibition in CEATg mice
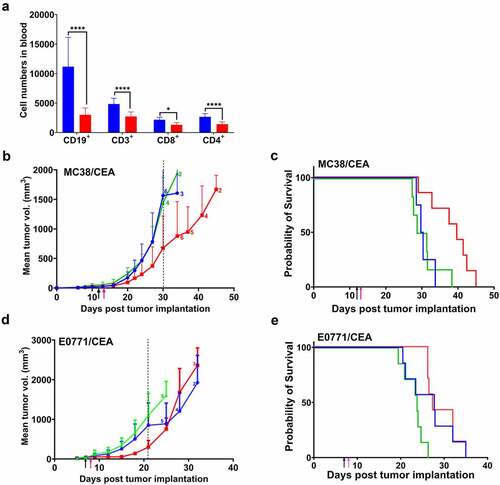
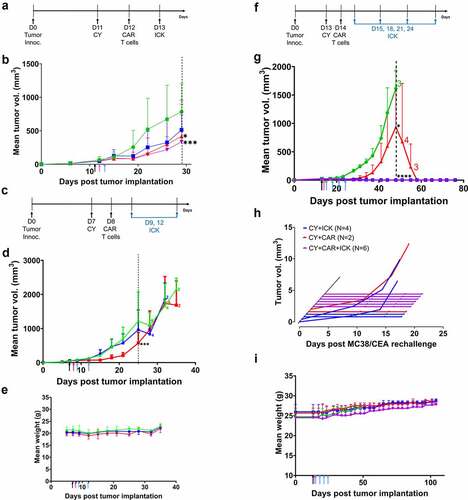
CY i.p. injected into CEA+ tumor-bearing, immunocompetent CEATg mice 24 hours prior to CAR T cell therapy depleted both B cells and T cells, 73% and 43%, respectively (). CY treatment combined with anti-CEA CAR T cell therapy delayed MC38/CEA tumor growth by 30 days compared to 24 days for anti-CEA CAR T cell therapy alone ( and Supplement ). The median overall survival in mice treated with CY plus anti-CEA CAR T cells had a statistically significant increase to 40 days compared to 30 days for mice treated with CY and mock T cells (p < .01) (). The median survival in mice treated with CY alone was also 30 days (). No whole-body toxicity was observed as measured by a decrease in mouse weight (Supplement ), as well as no diarrhea or loss of physical mobility. There were no morphology changes in CEA+ organs that were collected 3-days post T cell therapy and stained for human CEA (Supplement ), confirming the expression of CEA in the transgenic mice. Staining of the CEA+ tissues – collected 3 days post-T cell therapy (Supplement ) vs. the terminal timepoint of 1500 mm3 tumor size (Supplement ) – for murine CD3 revealed the presence of murine T cells in these tissues. In addition, there was no evidence of morphology changes in CEA+ organs at either the early or later timepoints, indicating lack of inflammatory tissue damage or presence of non-cytotoxic tissue infiltrating T cells.
Figure 3. CEA expression on s.c. MC38/CEA tumors in CEATg mice
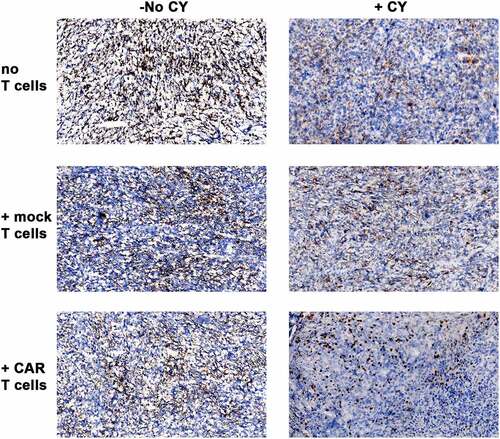
Figure 4. Therapeutic efficacy of cyclophosphamide plus repeated anti-CEA CAR T cell therapy on s.c. MC38/CEA tumor in CEATg mice
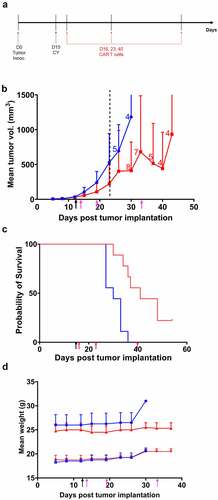
Figure 5. Effects of anti-CEA-IL2 immunocytokine (ICK) on anti-CEA CAR T cell activity in vitro.
To evaluate the efficacy of combined CY plus anti-CEA CAR T cell therapy in a more physiologically relevant tumor, orthotopic E0771/CEA breast adenocarcinoma tumor-bearing CEATg mice were treated with the combined treatments. The combined treatments delayed tumor growth by 21 days () compared to 30 days for the MC38/CEA tumors (). Mice bearing E0771/CEA tumors (N = 7) treated with CY and anti-CEA CAR T cells survived for 26 days post tumor implantation, while mice treated with CY alone or with CY plus Mock transduced T cells had a median survival of 19 days and 20 days, respectively (). No whole-body toxicity was observed as measured by a decrease in mouse weight (Supplement ), as well as no diarrhea or loss of physical mobility. These data suggest that CY improves antitumor activity of systemically delivered anti-CEA CAR T cells against both subcutaneous and orthotopic adenocarcinoma tumors without inducing off-target toxicity in CEATg mice.
Immune cell infiltrate phenotype analysis
To determine the effects of CY, MC38/CEA tumors were collected three days after CY plus either mock transduced T cell or anti-CEA CAR T cell therapy or at the termination of a study group (maximum tumor size of 1500 mm3). The collected tumors were sectioned and analyzed by immunohistochemistry to detect immune cell infiltration into tumor. Three days after CY and T cell treatments, high levels of CD3+ T cells, F4/80+ macrophages, and NKp46+ natural killer cells were found in tumors of all treatment groups (Supplement ). The highest levels of CD3+ T cells and F4/80+ macrophages were found in the tumors treated with CY plus anti-CEA CAR T cells (Supplement ). When tumors grew to the maximum size of 1500 mm3, there were much lower levels of CD3+ T cells, F4/80+ macrophages, and NKp46+ natural killer cells were found in tumors of all treatment groups (Supplement ). Interestingly, F4/80+ macrophages and NKp46+ natural killer cells detected at the termination timepoint were not evenly spread throughout the tumor compared to the 3-day post-therapy timepoint (Supplement ) but mostly found at the tumor edges or in streaks (Supplement ). Low levels of CD19+ B cells and few CD3+/CD19+ anti-CEA CAR T cells were found only in the 3-day post-therapy tumor treated with CY plus anti-CEA CAR T cells (Supplement ), evidence for CAR T cells that trafficked into the tumor. Once the tumor grew to 1500 mm3, no CD19+ B cells or CD3+/CD19+ anti-CEA CAR T cells were found in any of the tumors (Supplement ). There was evidence of Ly6G+ neutrophils only in 3 days post-therapy tumors treated with CY plus anti-CEA CAR T cells and none in tumors treated with any other treatments or in tumors at the terminal timepoint (Supplement and Supplement ).
Figure 6. Therapeutic efficacy of anti-CEA-IL2 immunocytokine (ICK) on CY plus anti-CEA CAR T cells on s.c. MC38/CEA and orthotopic E0771/CEA tumors in CEATg mice
Persistence of CEA expression after CY and T cell therapy
In hematological malignancies, antigen loss after CAR T cell therapy has been observed and correlated with relapse.Citation54–57 As result, CAR T cell therapy in these patients required therapy with a second tumor-associated antigen.Citation58,Citation59 To determine if CEA loss occurred in our CAR T therapy, MC38/CEA tumors were collected from mice with or without CY and/or CAR T cell therapy and then immunostained for CEA. CEA expression persisted in all MC38/CEA tumors after all treatments (). These data indicate that anti-CEA CAR T cell therapy can be repeatedly used to treat CEA+ tumors without cancer escape via antigen loss.
Pre-treatment with CY plus multiple anti-CEA CAR T cell therapy improves outcome
To prolong the antitumor activity of anti-CEA CAR T cell therapy, anti-CEA CAR T cell therapy was administered weekly for 3 weeks following CY treatment (). Two of 9 mice had complete eradication of MC38/CEA tumors after CY plus multiple anti-CEA CAR T cell therapies (). The median overall survival in mice treated with CY and multiple anti-CEA CAR T cell therapies was 41 days vs. 30 days in mice treated with CY plus mock transduced T cells (p < .001, ). There was no decrease in mouse weight (), or evidence of diarrhea and loss of physical mobility. These data suggest that, despite comparable overall survival between single and multiple anti-CEA CAR T cell therapies following CY treatment, multiple anti-CEA CAR T cell therapies may increase the incidence of tumor eradication without off-target toxicity in the CEATg mouse model.
Immunocytokine anti-CEA-IL2 increases the cytotoxic activity of anti-CEA CAR T cells
Although several anti-CEA CAR T cell clinical trials have added systemic IL2 to therapy to improve efficacy (NCT01373047; NCT02850536; NCT02416466; NCT03818165), patients were taken off IL2 treatment due to systemic IL2-related toxicity.Citation33 We and others have shown that systemic IL2 toxicity can be reduced by the use of targeted immunocytokines.Citation34,Citation60–62 Specifically, combined stereotactic radiation therapy (SRT) with anti-CEA-IL2 in the MC38/CEA and E0771/CEA tumor models in CEATg mice was found to not only stimulate a T cell-mediated antitumor response but also to lower regulatory T cell infiltration and stimulate a memory antitumor effect.Citation34 To determine if the addition of ICK to anti-CEA CAR T cell therapy would also improve the antitumor response, anti-CEA CAR or mock transduced T cells were co-cultured with MC38/GFP vs. MC38/CEA/GFP cells in in the presence or absence of anti-CEA-IL2 ICK. The anti-CEA CAR T cells with ICK exhibited a dose-dependent cytotoxic response to CEA+ vs. CEA− cells and secreted more IFNγ than anti-CEA CAR T cells alone or mock transduced T cells (-d). As expected, ICK co-cultured with mock transduced T cells also increased their cytotoxicity against target cells, but IFNγ secretion was not increased (). These data demonstrate that ICK increases the cytolytic activity of anti-CEA CAR T cells via enhancing antigen-specific T cell activation.
ICK treatment improves efficacy of anti-CEA CAR T cell therapy against subcutaneous and orthotopic CEA+ adenocarcinoma tumors in CEATg mice
To evaluate the effects of ICK on the efficacy of anti-CEA CAR T cells in vivo, MC38/CEA tumor-bearing CEATg mice were given an i.p. injection of ICK one day following the start of CY and anti-CEA CAR T cell therapy (). Combined treatment of CY and CAR T cells plus ICK therapy resulted in MC38/CEA tumor regression in 4/8 CEATg mice compared to 2/8 mice treated with CY and CAR T cells without ICK ( and Supplement Figure 7a-d).
Since a single ICK treatment added to CY plus anti-CEA CAR T cell therapy showed potential in improving the delay of tumor growth, two ICK injections were added to the therapy of the more aggressive E0771/CEA tumors (). Two ICK treatments, 3 days apart, added to the CY plus anti-CEA CAR T cell therapy delayed E0771/CEA tumor growth to 25 days compared to 21 days for mice treated with CY and anti-CEA CAR T cells alone (). As before, there was no decrease in mouse weights, or evidence of diarrhea or loss of physical mobility (). These data indicate that ICK enhances antitumor activity of anti-CEA CAR T cells against both MC38/CEA and E0771/CEA tumors without causing off-target toxicity in CEATg mice.
Lymphocytes in the tumors and tumor-draining lymph nodes (TDLNs) of the orthotopic E0771/CEA tumor model were analyzed by flow cytometry. Tissues were analyzed at the experimental end point of maximum allowed tumor volumes in which tumors relapsed. Nevertheless, mice treated with ICK had a significantly lower levels of tumor-infiltrating FoxP3+ regulatory T cells (Treg) than mice not treated with ICK, but the percent of FoxP3+ Treg cells was not affected by ICK in the TDLNs (Supplement Figure 8a). In all treatment groups, more CD4+ and CD8+ T cells were found in the TDLNs than in the tumors (Supplement Figure 8b and Supplement Figure 8c). Tumor-infiltrating CD8+ T cells were IFNγ+/PD1−, IFNγ+/PD1+, and IFNγ−/PD1+ but CD8+ T cells in TDLNs were predominantly IFNγ+/PD1− (Supplement Figure 8d). CD4+ T cells in the tumors and in the TDLNs were mostly IFNγ−/PD1+ (Supplement Figure 8e). These data indicate that CY plus anti-CEA CAR T cells combined with ICK predominantly recruit IFNγ+/PD1− CD8+ T cells from TDLNs into tumors and that ICK promotes a pro-inflammatory tumor microenvironment.
When the number of ICK treatments post CY plus CAR T cell therapy were increased to four times, three days apart, in the MC38/CEA tumor model (), tumor eradication was obtained in 6/6 treated mice compared to 2/6 in the CY plus anti-CEA CAR T cell group (). However, 4/4 mice treated with CY plus ICK also exhibited complete tumor eradication (), suggesting that CY plus 4 treatments of ICK alone was sufficient. To determine if CAR T therapy added benefit, mice with tumor regression were re-challenged with fresh tumor after 45 tumor-free days. Within seven days after the re-challenge, MC38/CEA tumors recurred in 0/6 of the CY plus anti-CEA CAR T cells plus ICK group, 2/4 of the CY plus ICK group, and 1/2 of the CY plus anti-CEA CAR T cells group (). As before, there was no decrease in mouse weight, no evidence of diarrhea, and no loss of physical mobility (). These data indicate that four ICK treatments following CY plus anti-CEA CAR T cell treatment was sufficient to not only eradicate MC38/CEA tumors but also to establish tumor immunity. Since the number of mice in the ICK only group were limited, further studies are required to determine the degree of immune protection provided by four treatments with ICK alone.
Discussion
CAR T cell therapy directed at CEA requires a careful selection of the starting antibody, since the CEA (CEACAM5) gene family includes a large number of cross-reacting antigens that are expressed in many normal tissues including lymphocytes.Citation63–66 In this regard, the last two domains of CEA, the A3B3 domains, are unique to CEA and not found in any of the CEACAM related genes.Citation67 Thus, we and others have selected A3B3 domain antibodies as a starting point. In addition, CAR T therapy against solid tumors presents a number of challenges that can be further addressed by approaches such as regional or combination therapy. For example, the efficacy of i.p. injected anti-CEA CAR T cells targeting the A3B3 domain of CEA was greater for i.p. vs. s.c. MC38/CEA tumors in a C57Bl/6 J mouse model, and the systemic IFNγ levels were higher with systemic IL2 plus anti-CEA CAR T therapy vs. systemic IL2 alone.Citation68 Further improvements were seen with strategies such as anti-PD1/PD-L1 therapy to reduce the anti-inflammatory tumor microenvironment.Citation68 However, those studies did not address the issue of CEA expression in the normal colon, since mice do not have an equivalent of the CEA gene. In follow-up clinical trials by the same investigators, systemic IL2 plus anti-CEA CAR T therapy had to be suspended due to severe toxicity.Citation33,Citation69,Citation70 The rationale for the addition of IL2 is to support CAR T cell survival and expansion in vivo.Citation71,Citation72 In another pre-clinical study with anti-CEA CAR T cells targeting the A3 domain of CEA, tumor regression was achieved in an orthotopic CEA+ pancreatic carcinoma tumor in CEA transgenic mice without inducing autoimmune colitis.Citation32 In an advanced tumor model study, second-generation anti-CEA CAR T cells were not sufficient for tumor regression prompting development of a fourth-generation version of anti-CEA CAR T cells that secreted IL18.Citation73 In that study, the addition of IL18 led to a proinflammatory tumor microenvironment with TbethighFoxO1low T cells. Taken together, these studies support the conclusion that CAR T therapy for solid tumors requires additional support with cytokine therapy delivered at the site of the tumor.
In our study, we selected the anti-CEA antibody T84.66 that is CEA specific and has been used in clinical assays for CEA.Citation74–76 In addition, the chimeric and humanized versions of this antibody have been used for in vivo imaging of CEA-positive tumors in the clinic with no evidence of targeting the normal colon.Citation24,Citation77–80 Since there is a distinct possibility that CAR T cells may target CEA in normal colon, in spite of their recognition of whole CEA rather than CEA peptides in the context of MHC, it was necessary to utilize a CEA transgenic mouse model allowing for CEA expression in normal tissues. In this respect, our CEATg mice express the entire human gene for CEA including its promoter and were shown to express CEA in the normal colon.Citation41 Our study with a second-generation CAR T construct directed at the A3 domain of CEA agrees with the findings of Abken and coworkers who showed a delay in CEA+ solid tumor growth without off-target toxicity using a second-generation CAR T construct directed at the A3 domain of CEA in a CEATg mouse.Citation32 Although the antitumor response was more effective in the pancreatic tumor model compared to our s.c. colon and orthotopic breast model, this difference was likely due to differential tumor sensitivity to therapy. Nonetheless, it was clear from both studies that additional therapy was required.
To improve the efficacy of anti-CEA CAR T cell monotherapy, we first modified the therapy with CY pre-treatment to make homeostatic space for the number of CAR T cells introduced. Along these lines, Parkhurst et al. found that nonmyeloablative lymphodepletion before CEA-reactive TCR T cell therapy was necessary in metastatic CEA+ melanoma patients for transduced T cell proliferation and persistence.Citation81–83 Kujawski et. al. also found that the addition of CY to ICK therapy depletes immunosuppressive regulatory T cells (Treg) cells.Citation34 Chmielewski et. al. found that lymphodepletion with CY (200 mg/kg) and fludarabine (150 mg/kg) before anti-CEA CAR T cell therapy did not cause autoimmune colitis, suggesting that CY was a safe method for lymphodepletion.Citation32 Our results support the previous studies in that CY pre-treatment improved the efficacy of CAR T cells without inducing normal tissue toxicity. Further improvements included multiple CAR T cell treatments to offset the rapid decline in circulating CAR T cells and the low percentage that reach the tumor.Citation37,Citation84 For example, in a CAR T cell study that targeted i.p. TAG72+ ovarian cancer in NSG mice, repeated i.p. injection of CAR T cells reduced tumor growth rate and extended overall survival compared to a single treatment strategy.Citation37 In our study, three weekly injections of anti-CEA CAR T cell therapy following CY resulted in complete tumor regression in 2/9 treated mice, but the tumor growth rate and median survival were comparable to CY and single anti-CEA CAR T cell therapy controls. Although further studies are needed to optimize the timing, number of injections, and dose to extend the antitumor response, these initial studies suggested that repeated CAR T therapy alone was insufficient for complete response.
To enhance the antitumor response of anti-CEA CAR T cells further, the addition of tumor-targeted ICK to CAR T cell therapy was explored. While it was shown that systemic IL2 therapy can enhance anti-CEA CAR T therapy in mice,Citation68 the same approach in man resulted in severe toxicity.Citation33 The use of an anti-CEA-IL2 ICK by Klein et al. without an CD25 binding domain demonstrated that ICK therapy specifically targeted CEA+ Panc02 tumor in CEATg mice and that when combined with anti-PD1 therapy enhanced the tumor response.Citation60 Recently, we have shown that our humanized anti-CEA-IL2 ICK combined with stereotactic radiation therapy eradicated orthotopic E0771/CEA tumors and established tumor immunity in a CEATg mouse model.Citation34 Thus, it was attractive to test this ICK in combination with CAR T cell therapy based on the same CEA epitope-specific antibody. In fact, CY and four ICK treatments with anti-CEA CAR T cells eradicated s.c. MC38/CEA tumors in 6/6 CEATg mice and rejected 6/6 tumors when re-challenged with fresh tumor.
Immunophenotyping of cells in tumors with or without treatments revealed the evidence of anti-CEA CAR T cells only in MC38/CEA tumors collected 3 days after CY plus anti-CEA CAR T cell therapy but none in MC38/CEA or E0771/CEA tumors collected at the maximum allowed size of 1500 mm3. These data indicate that tumor-infiltrating CAR T cells likely have exerted a direct tumor effect but did not persist long enough without assistance for tumor regression. In general, higher number of T cells, macrophages, and NK cells were found in MC38/CEA tumors treated with CY. This supported previous findings by Wu and Waxman that CY injections upregulated GL261 glioma tumor-infiltrating CD8+ T cells, NK cells, and macrophages and that the antitumor activities of these tumor-infiltrating immune cells are essential for tumor regression and memory in immune-competent mice.Citation85 We are unclear on the exact mechanism of CY-mediated effect on anti-CEA CAR T cell activity, but it is clear that CY promotes immune cell infiltration into MC38/CEA tumor to enhance tumor regression. In any case, the addition of ICK reduced the number of Treg cells in the tumor and created a proinflammatory tumor microenvironment in E0771/CEA tumors. Further studies are required to determine the exact mechanism of the amnestic response.
In terms of off-target toxicity, we did not observe significant weight loss or perturbations in RBCs, white cells, or platelets by the single or combined treatments. However, it is well known that mouse toxicity studies do not adequately predict toxicity in man. Thus, a safety trial in man is a logical next step. A second issue central to the use of ICK is that endogenous T cells will also respond since targeting of ICK to tumor is only in the range of 10% injected dose.Citation34 However, there may be a greater advantage for CAR T cells targeted to the tumor in that the local concentration of targeted ICK would be higher in the tumor than in the periphery. While it is also a possibility that ICK binding to CEA in the tumor would compete with target recognition by the CAR T cells, the low percentage of both targeted CAR T cells and ICK reaching the tumor (less than 5 and 10% of injected dose, respectively) compared to the number of CEA molecules per tumor cell (>105) makes this a minor consideration. This is evident in that one or two administrations of ICK were not effective until the dose was repeated four times. It should be noted that we have previously published on the effect of ICK alone in the absence of CY in the same animal model and ICK alone was insufficient to achieve complete tumor regression.Citation34 While it is too early to speculate on the mechanism of immune memory in the ICK only vs. ICK+CAR T groups without more data, it is likely that the population of endogenous antigen-specific T cells in the tumor is low and quiescent, and although they can respond to ICK, the result is a limited response with poor generation of memory cells. On the other hand, when very active antigen-specific CAR Ts arrive at the tumor, they may initiate an immune response that is amplified by ICK, not only enhancing the anti-CEA response but generating memory T cells in greater numbers. We have more work to do to determine if the response is CEA specific or if antigen spreading has occurred. At this time, we think the ICK plus CAR T memory response is based on absolute numbers of antigen-specific T cells at the tumor and/or lymph nodes.
Conclusion
We have shown that anti-CEA CAR T cell therapy is effective for both a s.c. colon and orthotopic breast cancer models and that it can be further optimized with prior lymphodepletion and the addition of ICK with no evidence of off-target effects in CEATg mice. Although further optimization of timing, dose, and frequency of treatments is envisioned, this study provides evidence that a clinical trial with CY, anti-CEA CAR T cells, and ICK is warranted.
Disclosure of interest
The authors report no conflict of interest.
Supplemental Material
Download ()Acknowledgments
We would like to thank Wen-Chung Chang at City of Hope for her assistance in designing constructs, Dr. John Murad at City of Hope for his insights on transduction methods, and City of Hope Pathology Core for IHC supported by NCI Cancer Center grant P30 CA033572.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- MayoClinic, Colon Cancer.
- Colon Cancer Treatment (PDQ(R)): Health Professional Version, in PDQ Cancer Information Summaries. 2002: Bethesda MD.
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–14. doi:10.3322/caac.21492.
- Sur D, Havasi A, Cainap C, Samasca G, Burz C, Balacescu O, Lupan I, Deleanu D, Irimie A. Chimeric antigen receptor t-cell therapy for colorectal cancer. J Clin Med. 2020;9(1):182-201. doi:10.3390/jcm9010182.
- Waks AG, Winer EP. Breast cancer treatment: a review. Jama. 2019;321(3):288–300. doi:10.1001/jama.2018.19323.
- Esteva FJ, Hubbard-Lucey VM, Tang J, Pusztai L. Immunotherapy and targeted therapy combinations in metastatic breast cancer. Lancet Oncol. 2019;20(3):e175–e186. doi:10.1016/S1470-2045(19)30026-9.
- Gold P, Freedman SO. Demonstration of tumor-specific antigens in human colonic carcinomata by immunological tolerance and absorption techniques. J Exp Med. 1965;121(3):439–462. doi:10.1084/jem.121.3.439.
- Molina R, Barak V, van Dalen A, Duffy MJ, Einarsson R, Gion M, Goike H, Lamerz R, Nap M, Sölétormos G, et al. Tumor markers in breast cancer- european group on tumor markers recommendations. Tumour Biol. 2005;26(6):281–293. doi:10.1159/000089260.
- Molina R, Jo J, Zanón G, Filella X, Farrus B, Muñoz M, Latre ML, Pahisa J, Velasco M, Fernandez P, et al. Utility of C-erbB-2 in tissue and in serum in the early diagnosis of recurrence in breast cancer patients: comparison with carcinoembryonic antigen and CA 15.3. Br J Cancer. 1996;74(7):1126–1131. doi:10.1038/bjc.1996.501.
- Molina R, Zanón G, Filella X, Moreno F, JJ, Daniels M, Latre ML, Giménez N, Pahisa J, Velasco M, et al. Use of serial carcinoembryonic antigen and CA 15.3 assays in detecting relapses in breast cancer patients. Breast Cancer Res Treat. 1995;36(1):41–48. doi:10.1007/BF00690183.
- Li X, Dai D, Chen B, Tang H, Xie X, Wei W. Determination of the prognostic value of preoperative CA15-3 and CEA in predicting the prognosis of young patients with breast cancer. Oncol Lett. 2018;16(4):4679–4688. doi:10.3892/ol.2018.9160.
- Eker B, Ozaslan E, Karaca H, Berk V, Bozkurt O, Inanc M, Duran AO, Ozkan M. Factors affecting prognosis in metastatic colorectal cancer patients. Asian Pac J Cancer Prev. 2015;16(7):3015–3021. doi:10.7314/APJCP.2015.16.7.3015.
- Powell E, Shao J, Picon HM, Bristow C, Ge Z, Peoples M, Robinson F, Jeter-Jones SL, Schlosberg C, Grzeskowiak CL, et al. A functional genomic screen in vivo identifies CEACAM5 as a clinically relevant driver of breast cancer metastasis. NPJ Breast Cancer. 2018;4:9. doi:10.1038/s41523-018-0062-x.
- Mansour EG, Hastert M, Park CH, Koehler KA, Petrelli M. Tissue and plasma carcinoembryonic antigen in early breast cancer. A prognostic factor. Cancer. 1983;51(7):1243–1248. doi:10.1002/1097-0142(19830401)51:7<1243::AID-CNCR2820510712>3.0.CO;2-A.
- Pietra N, Sarli L, Costi R, Ouchemi C, Grattarola M, Peracchia A. Role of follow-up in management of local recurrences of colorectal cancer: a prospective, randomized study. Dis Colon Rectum. 1998;41(9):1127–1133. doi:10.1007/BF02239434.
- Moertel CG, Fleming TR, Macdonald JS, Haller DG, Laurie JA, Tangen C. An evaluation of the carcinoembryonic antigen (CEA) test for monitoring patients with resected colon cancer. Jama. 1993;270(8):943–947. doi:10.1001/jama.1993.03510080047030.
- Graham RA, Wang S, Catalano PJ, Haller DG. Postsurgical surveillance of colon cancer: preliminary cost analysis of physician examination, carcinoembryonic antigen testing, chest x-ray, and colonoscopy. Ann Surg. 1998;228(1):59–63. doi:10.1097/00000658-199807000-00009.
- Woo J, Kim J, Park I, Cho H, Gwak G, Yang KH, Bae BN, Kim KH. Perioperative serum carcinoembryonic antigen ratio is a prognostic indicator in patients with stage ii colorectal cancer. Ann Coloproctol. 2018;34(1):4–10. doi:10.3393/ac.2018.34.1.4.
- Lin BR, Lin YL, Lai HS, Lee PH, Chang KJ, Liang JT. Overall survival of stage III colon cancer with only one lymph node metastasis is independently predicted by preoperative carcinoembryonic antigen level and lymph node sampling status. PLoS One. 2015;10(9):e0137053. doi:10.1371/journal.pone.0137053.
- Duffy MJ. Carcinoembryonic antigen as a marker for colorectal cancer: is it clinically useful? Clin Chem. 2001;47(4):624–630. doi:10.1093/clinchem/47.4.624.
- Duffy MJ, van Dalen A, Haglund C, Hansson L, Klapdor R, Lamerz R, Nilsson O, Sturgeon C, Topolcan O. Clinical utility of biochemical markers in colorectal cancer: European group on tumour markers (EGTM) guidelines. Eur J Cancer. 2003;39(6):718–727. doi:10.1016/S0959-8049(02)00811-0.
- Shousha S, Lyssiotis T, Godfrey VM, Scheuer PJ. Carcinoembryonic antigen in breast-cancer tissue: a useful prognostic indicator. Br Med J. 1979;1(6166):777–779. doi:10.1136/bmj.1.6166.777.
- Ordonez C, Screaton RA, Ilantzis C, Stanners CP. Human carcinoembryonic antigen functions as a general inhibitor of anoikis. Cancer Res. 2000;60(13):3419–3424.
- Li L, Bading J, Yazaki PJ, Ahuja AH, Crow D, Colcher D, Williams LE, Wong JYC, Raubitschek A, Shively JE. A versatile bifunctional chelate for radiolabeling humanized anti-CEA antibody with In-111 and Cu-64 at either thiol or amino groups: PET imaging of CEA-positive tumors with whole antibodies. Bioconjug Chem. 2008;19(1):89–96. doi:10.1021/bc700161p.
- Wong JY, Chu DZ, Williams LE, Liu A, Zhan J, Yamauchi DM, Wilczynski S, Wu AM, Yazaki PJ, Shively JE. A phase I trial of (90)Y-DOTA-anti-CEA chimeric T84.66 (cT84.66) radioimmunotherapy in patients with metastatic CEA-producing malignancies. Cancer Biother Radiopharm. 2006;21(2):88–100. doi:10.1089/cbr.2006.21.88.
- Wong JYC, Williams LE, Yamauchi DM, Odom-Maryon T, Esteban JM, Neumaier M, Wu AM, Johnson DK, Primus FJ, Shively JE, et al. Initial experience evaluating 90yttrium-radiolabeled anti-carcinoembryonic antigen chimeric T84.66 in a phase I radioimmunotherapy trial. Cancer Res. 1995;55(23 Supplement):5929s–5934s.
- Schlom J. Basic principles and applications of monoclonal antibodies in the management of carcinomas: the richard and hinda rosenthal foundation award lecture. Cancer Res. 1986;46:3225–3238.
- Kuroki M, Kuroki M, Koga Y, Matsuoka Y. Monoclonal antibodies to carcinoembryonic antigen: a systematic analysis of antibody specificities by using related normal antigens and evidence for allotypic determinants on carcinoembryonic antigen. J Immunol. 1984;133(4):2090–2097.
- Lonberg N, Taylor LD, Harding FA, Trounstine M, Higgins KM, Schramm SR, Kuo CC, Mashayekh R, Wymore K, McCabe JG, et al. Antigen-specific human antibodies from mice comprising four distinct genetic modifications. Nature. 1994;368(6474):856–859. doi:10.1038/368856a0.
- Primus FJ, Newell KD, Blue A, Goldenberg DM. Immunological heterogeneity of carcinoembryonic antigen: antigenic determinants on carcinoembryonic antigen distinguished by monoclonal antibodies. Cancer Res. 1983;43(2):686–692.
- Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16:566–581.
- Chmielewski M, Hahn O, Rappl G, Nowak M, Schmidt-Wolf IH, Hombach AA, Abken H. T cells that target carcinoembryonic antigen eradicate orthotopic pancreatic carcinomas without inducing autoimmune colitis in mice. Gastroenterology. 2012;143(4):1095–1107. e2. doi:10.1053/j.gastro.2012.06.037.
- Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, Khare PD, Thorn M, Ma Q, Stainken BF, et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res. 2015;21(14):3149–3159. doi:10.1158/1078-0432.CCR-14-1421.
- Kujawski M, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, Khare PD, Thorn M, Ma Q, Stainken BF et al. Potent immunomodulatory effects of an anti-CEA-IL-2 immunocytokine on tumor therapy and effects of stereotactic radiation. OncoImmunology. 2020;9(1):1724052. doi:10.1080/2162402X.2020.1724052.
- He Z, Ma J, Wang R, Zhang J, Huang Z, Wang F, Sen S, Rothenberg EV, Sun Z. A two-amino-acid substitution in the transcription factor RORgammat disrupts its function in TH17 differentiation but not in thymocyte development. Nat Immunol. 2017;18(10):1128–1138. doi:10.1038/ni.3832.
- Neumaier M, Shively L, Chen FS, Gaida FJ, Ilgen C, Paxton RJ, Shively JE. Cloning of the genes for T84.66, an antibody that has a high specificity and affinity for carcinoembryonic antigen, and expression of chimeric human/mouse T84.66 genes in myeloma and Chinese hamster ovary cells. Cancer Res. 1990;50(7):2128–2134.
- Murad JP, Kozlowska AK, Lee HJ, Ramamurthy M, Chang WC, Yazaki P, Colcher D, Shively J, Cristea M, Forman SJ, et al. Effective targeting of TAG72(+) peritoneal ovarian tumors via regional delivery of CAR-engineered t cells. Front Immunol. 2018;9:2268. doi:10.3389/fimmu.2018.02268.
- Brown CE, Aguilar B, Starr R, Yang X, Chang W-C, Weng L, Chang B, Sarkissian A, Brito A, Sanchez JF, et al. Optimization of IL13Rα2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Molecular Therapy. 2018;26(1):31–44. doi:10.1016/j.ymthe.2017.10.002.
- Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK, Chang WC, Ostberg JR, Neman J, Jandial R, et al. Regional delivery of chimeric antigen receptor-engineered T cells effectively targets HER2(+) breast cancer metastasis to the brain. Clin Cancer Res. 2018;24(1):95–105. doi:10.1158/1078-0432.CCR-17-2041.
- Donnelly MLL, Luke G, Mehrotra A, Li X, Hughes LE, Gani D, Ryan MD. Analysis of the aphthovirus 2A/2B polyprotein ‘cleavage’ mechanism indicates not a proteolytic reactionbut a novel translational effect: a putative ribosomal ‘skip’. J Gen Virol. 2001;82(Pt 5):1013–1025.
- Clarke P,Mann J, Simpson JF, Rickard-Dickson K, Primus FJ. Mice transgenic for human carcinoembryonic antigen as a model for immunotherapy. Cancer Res. 1998;58(7):1469–1477.
- Yazaki PJ, Wu AM, Tsai SW, Williams LE, Ikle DN, Wong JYC, Shively JE, Raubitschek AA. Tumor targeting of radiometal labeled anti-cea recombinant t84.66 diabody and t84.66 minibody: Comparison to radioiodinated fragments. Bioconjug Chem. 2001;12(2):220–228. doi:10.1021/bc000092h.
- Crouch ML,Knowels G, Stuppard R, Ericson NG, Bielas JH, Marcinek DJ, Syrjala KL. Cyclophosphamide leads to persistent deficits in physical performance and in vivo mitochondria function in a mouse model of chemotherapy late effects. PLoS One. 2017;12(7):e0181086. doi:10.1371/journal.pone.0181086.
- Jafarzadeh L,Masoumi E, Fallah-Mehrjardi K, Mirzaei HR, Hadjati J. Prolonged persistence of chimeric antigen receptor (CAR) T cell in adoptive cancer immunotherapy: Challenges and ways forward. Front Immunol. 2020;11:702. doi:10.3389/fimmu.2020.00702.
- Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202(7):907–912. doi:10.1084/jem.20050732.
- Bracci L, oschella F, Sestili P, La Sorsa V, Valentini M, Canini I, Baccarini S, Maccari S, Ramoni C, Belardelli F, et al. Cyclophosphamide enhances the antitumor efficacy of adoptively transferred immune cells through the induction of cytokine expression, B-cell and T-cell homeostatic proliferation, and specific tumor infiltration. Clin Cancer Res. 2007;13(2 Pt 1):644–653. doi:10.1158/1078-0432.CCR-06-1209.
- Xu J, Wang Y, Shi J, Liu J, Li Q, Chen L. Combination therapy: a feasibility strategy for CAR-T cell therapy in the treatment of solid tumors. Oncol Lett. 2018;16(2):2063–2070. doi:10.3892/ol.2018.8946.
- Vierboom MP,Bos GM, Ooms M, Offringa R, Melief CJ. Cyclophosphamide enhances anti-tumor effect of wild-type p53-specific CTL. Int J Cancer. 2000;87(2):253–260. doi:10.1002/1097-0215(20000715)87:2<253::AID-IJC17>3.0.CO;2-A.
- Alizadeh D,Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, Katsanis E, Larmonier N. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 2014;74(1):104–118. doi:10.1158/0008-5472.CAN-13-1545.
- Peng KW,Myers R, Greenslade A, Mader E, Greiner S, Federspiel MJ, Dispenzieri A, Russell SJ. Using clinically approved cyclophosphamide regimens to control the humoral immune response to oncolytic viruses. Gene Ther. 2013;20(3):255–261. doi:10.1038/gt.2012.31.
- Lutsiak ME,Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood. 2005;105(7):2862–2868. doi:10.1182/blood-2004-06-2410.
- Myers CE,Hoelzinger DB, Truong TN, Chew LA, Myles A, Chaudhuri L, Egan JB, Liu J, Gendler SJ, Cohen PA. Chemotherapy can induce weight normalization of morbidly obese mice despite undiminished ingestion of high fat diet. Oncotarget. 2017;8(3):5426–5438. doi:10.18632/oncotarget.14576.
- Eftekhar E, Naghibalhossaini F. Carcinoembryonic antigen expression level as a predictive factor for response to 5-fluorouracil in colorectal cancer. Mol Biol Rep. 2014;41(1):459–466. doi:10.1007/s11033-013-2880-0.
- Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8(10):1219–1226.
- Maude SL, Teachey DT, Rheingold SR, Shaw PA, Aplenc R, Barrett DM, Barker CS, Callahan C, Frey NV, Nazimuddin F, et al. 2016. Sustained remissions with CD19-specific chimeric antigen receptor (CAR)-modified T cells in children with relapsed/refractory ALL. Vol. 34( 15_suppl): 3011.
- Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, Sussman R, Lanauze C, Ruella M, Gazzara MR, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5(12):1282–1295. doi:10.1158/2159-8290.CD-15-1020.
- Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, Levine JE, Qayed M, Grupp SA, Boyer M, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. 2018;24(10):1504–1506. doi:10.1038/s41591-018-0146-z.
- Shalabi H, Kraft IL, Wang HW, Yuan CM, Yates B, Delbrook C, Zimbelman JD, Giller R, Stetler-Stevenson M, Jaffe ES, et al. Sequential loss of tumor surface antigens following chimeric antigen receptor T-cell therapies in diffuse large B-cell lymphoma. Haematologica. 2018;103(5):e215–e218. e215-e218. doi:10.3324/haematol.2017.183459.
- Qin H, Dong Z, Wang X, Cheng WA, Wen F, Xue W, Sun H, Walter M, Wei G, Smith DL, et al. CAR T cells targeting BAFF-R can overcome CD19 antigen loss in B cell malignancies. Sci Transl Med. 2019;11(511):eaaw9414. doi:10.1126/scitranslmed.aaw9414.
- Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, Dunn C, Bolijn M, Benz J, Stihle M, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology. 2017;6(3):e1277306. doi:10.1080/2162402X.2016.1277306.
- Ribba B, Boetsch C, Nayak T, Grimm HP, Charo J, Evers S, Klein C, Tessier J, Charoin JE, Phipps A, et al. Prediction of the optimal dosing regimen using a mathematical model of tumor uptake for immunocytokine-based cancer immunotherapy. Clin Cancer Res. 2018;24(14):3325–3333. doi:10.1158/1078-0432.CCR-17-2953.
- Van Brummelen EMJ, Huisman MC, de Wit-van der Veen LJ, Nayak TK, Stokkel MPM, Mulder ER, Hoekstra OS, Vugts DJ, Van Dongen G, Verheul HM, et al. (89)Zr-labeled CEA-targeted IL-2 variant immunocytokine in patients with solid tumors: CEA-mediated tumor accumulation and role of IL-2 receptor-binding. Oncotarget. 2018;9(37):24737–24749. doi:10.18632/oncotarget.25343.
- Shively JE, Beatty JD. CEA-related antigens: molecular biology and clinical significance. Crit Rev Oncol Hematol. 1985;2:355–399.
- Hammarstrom S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol. 1999;9(2):67–81. doi:10.1006/scbi.1998.0119.
- Coutelier JP, Godfraind C, Dveksler GS, Wysocka M, Cardellichio CB, Noël H, Holmes KV. B lymphocyte and macrophage expression of carcinoembryonic antigen-related adhesion molecules that serve as receptors for murine coronavirus. Eur J Immunol. 1994;24(6):1383–1390. doi:10.1002/eji.1830240622.
- Kammerer R, Hahn S, Singer BB, Luo JS, von Kleist S. Biliary glycoprotein (CD66a), a cell adhesion molecule of the immunoglobulin superfamily, on human lymphocytes: structure expression and involvement in T cell activation. Eur J Immunol. 1998;28(11):3664–3674.
- Tchoupa AK, Schuhmacher T, Hauck CR. Signaling by epithelial members of the CEACAM family - mucosal docking sites for pathogenic bacteria. Cell Commun Signal. 2014;12(1):27. doi:10.1186/1478-811X-12-27.
- Katz SC, Point GR, Cunetta M, Thorn M, Guha P, Espat NJ, Boutros C, Hanna N, Junghans RP. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. 2016;23(5):142–148. doi:10.1038/cgt.2016.14.
- SorrentoTherapeutics I. Sorrento Therapeutics anti-CEA CAR-t demonstrates promising clinical activity and safety in phase Ib clinical trial. 2017.
- SorrentoTherapeutics I. Sorrento therapeutics autologous anti-CEA CAR-T cell therapy for liver metastases demonstrates therapeutic activity in stage iv pancreas cancer in a phase 1b HITM-SURE Trial (NCT02850536). GlobeNewswire; 2018.
- Lo AS, Lo AS, Ma Q, Liu DL, Junghans RP. Anti-GD3 chimeric sFv-CD28/T-cell receptor zeta designer T cells for treatment of metastatic melanoma and other neuroectodermal tumors. Clin Cancer Res. 2010;16(10):2769–2780. doi:10.1158/1078-0432.CCR-10-0043.
- Junghans RP, Ma Q, Rathore R, Gomes EM, Bais AJ, Lo ASY, Abedi M, Davies RA, Cabral HJ, Al-Homsi AS, et al. Phase I Trial of Anti-PSMA Designer CAR-T Cells in Prostate Cancer: possible Role for Interacting Interleukin 2-T Cell Pharmacodynamics as a Determinant of Clinical Response. Prostate. 2016;76(14):1257–1270.
- Chmielewski M, Abken H. CAR T cells releasing IL-18 Convert to T-Bet(high) FoxO1(low) effectors that exhibit augmented activity against advanced solid tumors. Cell Rep. 2017;21(11):3205–3219. doi:10.1016/j.celrep.2017.11.063.
- Wagener C, Clark BR, Rickard KJ, Shively JE. Monoclonal antibodies for carcinoembryonic antigen and related antigens as a model system: determination of affinities and specificities of monoclonal antibodies by using biotin-labeled antibodies and avidin as precipitating agent in a solution phase immunoassay. J Immunol. 1983;130(5):2302–2307.
- Esteban JM, Felder B, Ahn C, Simpson JF, Battifora H, Shively JE. Prognostic relevance of carcinoembryonic antigen and estrogen receptor status in breast cancer patients. Cancer. 1994;74(5):1575–1583. doi:10.1002/1097-0142(19940901)74:5<1575::aid-cncr2820740513>3.0.co;2-w.
- Shively JE, Spayth V, Chang FF, Metter GE, Klein L, Presant CA, Todd CW. Serum levels of carcinoembryonic antigen and a tumor-extracted carcinoembryonic antigen-related antigen in cancer patients. Cancer Res. 1982;42(6):2506.
- Beatty JD, Duda RB, Williams LE, Sheibani K, Paxton RJ, Beatty BG, Philben VJ, Werner JL, Shively JE, Vlahos WG, et al. Preoperative imaging of colorectal carcinoma with 111in-labeled anticarcinoembryonic antigen monoclonal antibody. Cancer Res. 1986;46(12 Part 1):6494–6502.
- Yazaki PJ, Lee B, Channappa D, Cheung CW, Crow D, Chea J, Poku E, Li L, Andersen JT, Sandlie I, et al. A series of anti-CEA/anti-DOTA bispecific antibody formats evaluated for pre-targeting: comparison of tumor uptake and blood clearance. Protein Eng Des Sel. 2013;26(3):187–193. doi:10.1093/protein/gzs096.
- Nittka S, Krueger MA, Shively JE, Boll H, Brockmann MA, Doyon F, Pichler BJ, Neumaier M. Radioimmunoimaging of liver metastases with PET using a 64Cu-labeled CEA antibody in transgenic mice. PLoS One. 2014;9(9):e106921. doi:10.1371/journal.pone.0106921.
- Wu AM, Yazaki PJ, Tsai S, Nguyen K, Anderson AL, McCarthy DW, Welch MJ, Shively JE, Williams LE, Raubitschek AA, et al. High-resolution microPET imaging of carcinoembryonic antigen-positive xenografts by using a copper-64-labeled engineered antibody fragment. Proc Natl Acad Sci U S A. 2000;97(15):8495–8500. doi:10.1073/pnas.150228297.
- Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19(3):620–626. doi:10.1038/mt.2010.272.
- Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850–854. doi:10.1126/science.1076514.
- Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, Huang J, Powell DJ, Jr., Rosenberg SA. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173(12):7125–7130. doi:10.4049/jimmunol.173.12.7125.
- Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, Lin AA, Schlom J, June CH, Sherwin SA. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. 2017;5(1):22. doi:10.1186/s40425-017-0222-9.
- Wu J, Waxman DJ. Metronomic cyclophosphamide eradicates large implanted GL261 gliomas by activating antitumor Cd8(+) T-cell responses and immune memory. Oncoimmunology. 2015;4(4):e1005521. doi:10.1080/2162402X.2015.1005521.