
ABSTRACT
Chimeric antigen receptor (CAR)-T therapy holds great promise to sustainably improve cancer treatment. However, currently, a broad applicability of CAR-T cell therapies is hampered by limited CAR-T cell versatility and tractability and the lack of exclusive target antigens to discriminate cancerous from healthy tissues. To achieve temporal and qualitative control on CAR-T function, we engineered the Adapter CAR (AdCAR) system. AdCAR-T are redirected to surface antigens via biotin-labeled adapter molecules in the context of a specific linker structure, referred to as Linker-Label-Epitope. AdCAR-T execute highly specific and controllable effector function against a multiplicity of target antigens. In mice, AdCAR-T durably eliminate aggressive lymphoma. Importantly, AdCAR-T might prevent antigen evasion by combinatorial simultaneous or sequential targeting of multiple antigens and are capable to identify and differentially lyse cancer cells by integration of adapter molecule-mediated signals based on multiplex antigen expression profiles. In consequence the AdCAR technology enables controllable, flexible, combinatorial, and selective targeting.
One Sentence Summary
Adapter CAR-T cells for multiple synchronic targeting.
Introduction
CAR-T therapy has demonstrated durable clinical responses in patients with hematologic malignancies and raised great expectations for treatment of other cancer entities. Complete remission rates were achieved in patients suffering from B-lineage malignancies with 54% in a patient group of refractory diffuse large B-cell lymphoma, primary mediastinal B-cell lymphoma, or transformed follicular lymphomaCitation1 and 90% in refractory pediatric B-lineage derived ALL,Citation2 which led to FDA approval of CD19-CAR-T therapeutics.Citation1,Citation3 Despite these successes, various limitations of conventional CAR design appear when translating the technology for a broader spectrum of indications. Conventional CARs are engineered receptors consisting of an extracellular binding domain linked via a transmembrane domain to intracellular costimulatory and signaling domains (). Despite great potency, conventional CARs can only transmit an on- (presence of antigen) or off-signal (absence of antigen). The consequences are: (i) after initial antigen encounter in the patient, CAR-T expand massively in a potentially uncontrolled manner, which may cause life-threatening cytokine release syndrome and neurotoxicity;Citation4–6 (ii) effective CAR-T may lead to prolonged depletion of normal tissues expressing the target antigen, that can be life-threatening if the antigen is expressed on vitally essential tissue;Citation3 (iii) continuous stimulation drives CAR-T into exhaustion limiting efficacy; (iv) monovalent CAR-T create a strong selection pressure on the tumor, leading to the selection of target antigen low or negative tumor cells. Antigen loss is a major cause of treatment failureCitation7–10 and thus monotargeting is a key limitation von conventional CAR-T. Especially, cancers with heterogeneous antigen expression are likely to fail single-targeted therapy.Citation11 Addressing these limitations, novel CAR designs should combine controllability, flexibility and selectivity. Controllability should allow a tunable “ON”-state, secure and reversible “OFF”-state as well as temporal on-/off- switching, flexibility should allow multiple synchronic as well as sequential targeting and selectivity should enable a safe discrimination of cancerous versus healthy cells, e.g. by incorporating pharmacological switchesCitation12 or drug-inducible CAR expression.Citation13
Figure 1. Design and characterization of the AdCAR-T system
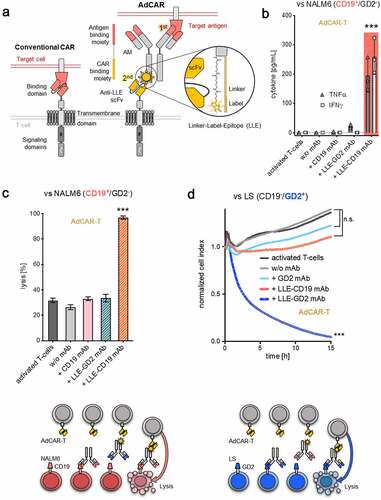
One elegant way to combine several essential features is to split target antigen recognition and CAR-T activation by introducing adapter molecules (AMs). AMs, defined by two functional moieties, an antigen binding moiety and a CAR binding moiety, can reversibly redirect CAR-T to different tumor-associated antigens (TAA).Citation14–23 Here, we report on the development of a novel anti-tag CAR-T system, referred to as AdCAR-T. We show that AdCAR-T mediate potent and specific antitumor activity against a multiplicity of TAAs in various cancer entities in vitro and in vivo. We demonstrate a CAR-technology that facilitates universal targeting, finely tunable effector function, mitigation of immune evasion due to antigen loss and differential identification and elimination of cancerous cells based on the integration of multiplex antigen expression profiles.
Results
Design of the AdCAR-T system
AdCAR-T comprises a two-component signal transduction system based on a split recognition/activation design in which labeled AMs are applied to transmit antigen recognition into T-cell activation via an anti-label CAR. AdCAR-T mediated T-cell activation and target-cell lysis is the result of a two-step process (), i) antigen-specific binding of the AM on the target cell and ii) binding of the AdCAR-expressing T cell to the AM. We used mAbs and mAb fragments as AMs to utilize their specific surface antigen-binding capacity. AMs are generated by biotinylation using a specific linker chemistry, resulting in a molecule comprising an antigen-binding moiety, a linker moiety and a label moiety (biotin) (). The AdCAR is based on unique characteristics of the mAb mBio3. mBio3 binds to biotin in the context of a specific linker, referred to as a Linker-Label Epitope (LLE). We designed AdCAR-T by conjugating the murine clone mBio3-derived scFvs on seond- and third-generation CAR backbones consisting of different extracellular spacer domains, as indicated, CD8 transmembrane, 4–1BB or CD28 and 4–1BB costimulatory as well as CD3-ζ signaling domain. Truncated human low-affinity nerve growth factor receptor (ΔLNGFR) was integrated downstream of a F2A site for detection and enrichment. In vitro testing was performed using the AdCAR-4–1BB-CD3-zeta, IgG4 hinge only, construct if not annotated otherwise. For initial evaluation, we LLE-conjugated the Fc-optimized CD19–4 G7SDIE mAb (LLE-CD19 mAb) and the FDA/EMA-approved GD2 mAb ch14.18/dinutuximab beta (LLE-GD2 mAb), both in clinical use at our institution.Citation24,Citation25
AdCAR-T mediate highly specific target-cell lysis
AdCAR can be expressed on activated T cells by lentiviral transduction as determined by ΔLNGFR expression (fig. S1A). The applied expansion protocol consistently resulted in balanced CD4+ to CD8+ T-cell ratios (fig. S1B). AdCAR-T got activated exclusively in the presence of antigen-specific LLE-conjugated AMs and the corresponding antigen-expressing target cell, assessed by cytokine production and transition into an effector phenotype (, fig. S1C). Importantly, AdCAR-T activation resulted in specific LLE-AM-mediated target-cell lysis, demonstrated for LLE-CD19 mAb and LLE-GD2 mAb against the CD19+ BCP-ALL cell line NALM6 and the GD2+ neuroblastoma cell line LS. No specific lysis was observed in the presence of antigen-specific mAb without LLE conjugation or LLE-AMs of irrelevant specificity (, C-D). Target-cell lysis was determined by LCA or by xCELLigence an impedance-based real-time cytotoxicity assay (ICA), that provides a real-time analysis. The molecular architecture of the different AdCAR variants is indicated in (fig. S2A). In these experiments the molecular variation was limited to the spacer length. A similar AdCAR expression level on T cells for the different AdCAR constructs is shown after LNGFR enrichment by microbeads in (fig. S2B). Comparing the impact of different extracellular spacer domains,Citation21,Citation26,Citation27 we found superior performance of the short, IgG4-hinge (IgG4S) construct independent of the i) target antigen CD19 in BCP-ALL targeting NALM6 (fig. S2C) or GD2 in neuroblastoma targeting LS (fig. 2SE) and independent of the ii) the AM format, full-size LLE-CD19 mAb or fragments thereof [LLE-CD19 F(ab’)2, LLE-Fab], despite the different AM-size (fig. S2D) with regard to cytotoxicity (fig. S2, C-E) and cytokine production (fig. S2F). Consequently, the AdCAR construct with the IgG4-hinge spacer was used for all subsequent experiments. In vitro testing revealed no differences in effector function of second- versus third-generation AdCAR constructs in terms of cytotoxicity and repetitive cytotoxicity (fig. S3, A-B), however with regard to proliferative capacity, third-generation AdCAR-T were superior (fig. S3C). The extracellular domain of the second- and third-generation AdCAR construct was the scFv in heavy-light configuration with an IgG4-hinge spacer.
AdCAR-T function is not impaired in the presence of biotin
One major obstacle of using biotin as a tag is the presence of free and protein-bound biotin in the human body. To test possible interference with free or protein-bound biotin, we assessed LLE-AM mediated target-cell lysis by AdCAR-T under increasing concentrations of biotin, up to 10000-fold of the physiological concentration,Citation17 or in the presence of 50% human serum. Strikingly, there was only minor inhibition of AdCAR-T function at supraphysiologic free biotin concentrations, further underscoring the selective binding of the AdCAR to the LLE-tag (, fig. S4B). Moreover, AdCAR-T are not activated unspecifically by oligobiotinylated LLE-CD19 mAb (fig. S12A). The dose of labeling was in the range of two to three biotins per antibody molecule as measured by mass spectrometry (fig. S12B).
Figure 2. Specificity and sensitivity of the AdCAR-T system
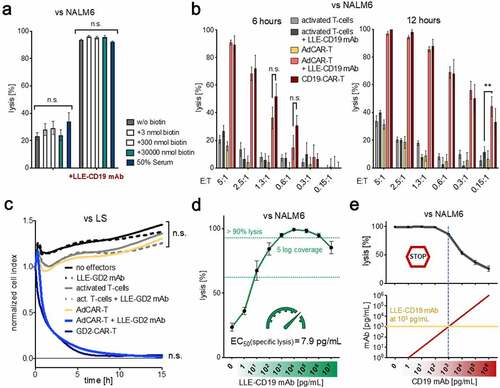
AdCAR-T mediate highly efficient and tightly controllable target-cell lysis
AdCAR-T sufficiently lysed target cells in vitro even at low E:T ratios (1:10), demonstrating serial killing capacity comparable to conventional CD19- and GD2-CAR-T cells (, B-C). Target-cell lysis was determined by LCA or by xCELLigence an impedance-based real-time cytotoxicity assay (ICA), that provides a real-time analysis. Lysis was equally mediated by biotinylated full-size LLE-mAb, LLE-F(ab’)2 and LLE-Fab (fig. S2D). AdCAR-T efficiently lysed target cells at LLE-AM concentrations starting as low as 1 pg/mL (EC50 of LLE-CD19 mAb vs. NALM6 = 7.9 pg/mL) with a coverage of 5 log-levels at highly potent activity, ensuring a wide therapeutic range with optimal effector function > 1 ng/mL LLE-AM (, fig. S4A). LLE-AM titration enabled finely tunable “ON”-switch function. Moreover, LLE-AM dependent AdCAR-T activity could also be terminated by addition of antigen-specific mAbs without LLE-tag, functioning as an “OFF”-switch (), underscoring the specificity and controllability of AdCAR-T system.
AdCAR-T eradicate disseminated lymphoma in vivo
Having proven functionality of the AdCAR system in vitro, we next set out to investigate efficacy in vivo. First, we tested the general in vitro stability of the LLE-conjugation of full length mAb in human whole blood for 24 hours (). Then we evaluated mouse plasma LLE-mAb levels 24 h after intravenous (iv) versus intraperitoneal (ip) application of 5 µg LLE-mAb and found no clear benefit of iv-application of the LLE-mAb compared to ip-application (). Consequently, we proceeded with ip-application of LLE-mAb in NSG mice in all subsequent in vivo experiments. The plasma levels of LLE-CD19 mAb were measured after 1 week and after 2 weeks in nontumor bearing mice () and revealed a plasma level half-life of t1/2 = 3.19 days at 50 µg ip application of LLE-CD19 mAb (fig. S4C). Consequently, we applied LLE-mAb twice per week. Further, we tested the impact of coadministration of 10 mg human IgG ip to saturate Fc-receptors and thus to mitigate Fc-receptor-mediated effector function. There was no impact of LLE-mAb mouse plasma levels without hIgG compared to the condition with coadministration of 10 mg hIgG (fig. S4D).
Figure 3. In vivo evaluation of AdCAR-T
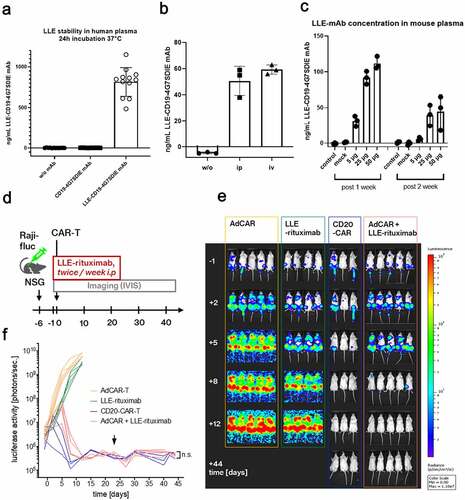
In vitro functional testing elucidated a significant higher proliferative capacity of third-generation AdCAR-T compared to second-generation AdCAR-T (fig. S3C) and in vivo comparison of second- versus third-generation AdCAR-T confirmed the importance of CAR-T cell proliferation to eradicate leukemia in a NALM6 tumor model utilizing the LLE-CD19 4G7SDIE mAb as AM. The superiority of third- versus second-generation CAR systems in adapter CAR technologies has also been previously reported by other groups.Citation28 Conventional CD19 CAR-T cells served as positive controls and cleared NALM6 from NSG mice more rapidly than the third-generator AdCAR-T (fig. S3D). In consequence, for subsequent in vivo evaluation of the AdCAR technology we utilized the third-generation AdCAR-CD28–4-1BB-CD3-zeta construct.
To demonstrate the operational versatility and the potential for straight clinical translation of the AdCAR-T technology, we chose the FDA/EMA approved CD20 mAb rituximab for in vivo evaluation. Rituximab was LLE-conjugated, achieving a purity of >95% LLE-rituximab (average 2 LLE/mAb). AdCAR-T were generated on the CliniMACS ProdigyTM, allowing a GMP-compliant closed-system processing for cell enrichment, activation, transduction, washing, and expansion.Citation29,Citation30 To underscore in vivo efficacy, we decided to use a rapidly progressive xenograft model of Burkitt’s lymphoma (Raji cell line) (, fig. S5A).Citation29 After confirmation of homogeneous engraftment on day −1 (fig. S5B), mice were grouped and the treatment according to indicated conditions was initiated. LLE-rituximab was injected intraperitoneally at 50 µg twice weekly starting on day −1 and suspended on day +23. AdCAR-T were injected intravenously on day 0, 6.23 × 106 AdCAR-T and a total of 20 × 106 human T cells per mouse (). Conventional CD20-CAR-T,Citation29 6.07 × 106 cells per mouse, served as a positive control. We did not observe signs of GvHD in the mouse models. Tumor burden was assessed by in vivo bioluminescence imaging (BLI). Strikingly, AdCAR-T in combination with LLE-rituximab completely eradicated disseminated lymphoma, as efficient as conventional CD20-CAR-T, although with slightly delayed kinetics. Mice remained in complete remission, demonstrated by BLI and flow cytometry of bone marrow, even after LLE-rituximab administration was terminated. In contrast, neither AdCAR-T nor LLE-rituximab alone mediated a significant effect on tumor burden (, E-F, fig. S5A). Solid engraftment of AdCAR-T was confirmed in the bone marrow on day +44 (fig. S5C).
In a NHL JeKo-1 tumor model 1 × 107 AdCAR-T were challenged to control and eradicate tumor burden at two different dose levels of either LLE-CD19 4G7SDIE mAb or LLE-CD20 rituximab applied ip twice per week (DL1 50 µg, DL2 5 µg). AdCAR-T plus CD19 mAb and CD20 mAb without LLE conjugation served as negative controls. At DL1 either with LLE-CD19 mAb or LLE-CD20 mAb, AdCAR-T controlled tumor growth, whereas at DL2 tumor growth kinetics were delayed in 3 mice and one mouse only achieved tumor clearance (fig. S3, E-F). Collectively, these data clearly underscore high in vivo therapeutic efficacy of the AdCAR-T system and warrant that clinical investigation should be initiated.
AdCAR-T mediate target-cell lysis against a variety of antigens proportionately to the frequency of antigen positive expressing target cells
One key feature of the adapter approach is the possibility for versatile and universal targeting. To test this hypothesis, we generated LLE-mAbs targeting antigens, associated with myeloid malignancies: CD15, CD32, CD33, CD38, CD117, CD123, CD133, CD135, CD276, and CD371. These antigens were chosen based on literature search and due to confirmation of substantial expression in AML cell lines that were immunophenotyped in our lab for a proof-of-concept.
LLE-mAb specific target-cell lysis was evaluated in the acute myeloid leukemia (AML) cell lines HL-60 and Kasumi-1, harboring inter- and intratumoral heterogeneity of target antigen expression. We found highly efficient target-cell lysis for 9 out of 10 evaluated LLE-AMs. Importantly, the percentage of specific lysis correlated well with the percentage of antigen-expressing cells in general, indicating the ability to specifically eliminate antigen-positive cells, while sparing antigen-negative cells (, A-B).
Figure 4. Universal targeting and combinatorial targeting to avoid antigen evasion
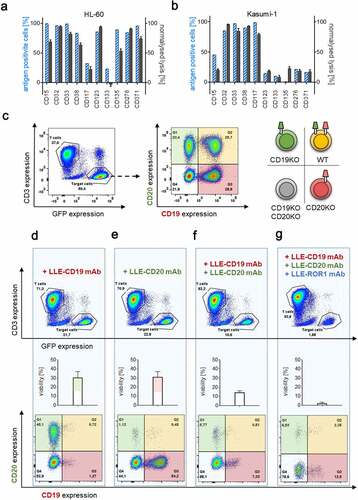
Simultaneous or sequential targeting might prevent selection of antigen-loss variants
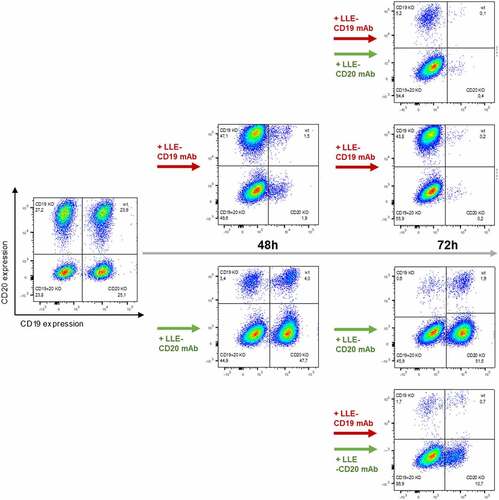
Therapy failure due to antigen evasion is one of the major clinical challenges in CAR-T therapy.Citation7,Citation8 To address this issue, we generated antigen-loss variants for CD19, CD20, and CD19/CD20 of the non-Hodgkin Lymphoma (NHL) cell line JeKo-1 by CRISPR/Cas9 knockout. All JeKo-1 variants showed similar growth kinetics, allowing simultaneous evaluation (fig. S6A). To test, whether dual or multiple targeting might prevent the specific selection of antigen-loss variants, we exposed AdCAR-T with a mix of wild type and knockout variants (, fig. S6B). Single targeting by either LLE-CD19 mAb or LLE-CD20 mAb eliminated target antigen positive cells with high selectivity, while sparing antigen negative variants (, D-E, fig. S6, C-D). Moreover, dual targeting led to the selection of double negative Jeko-1 cells (, fig. S6E). Addition of a 3rd AM, LLE-ROR-1 mAb, led to additional reduction of the double negative subset underscoring the capability for multiple “OR”-gating by AdCAR-T (). Monotargeting using the LLE-ROR-1 mAb significantly reduced the cell number in all four JeKo-1 variants comparably (fig. S6F). Moreover, AdCAR-T were demonstrated to be capable and flexible in sequential targeting. Thus sequential use of LLE-mAbs of different specificity () led to the anticipated reduction of the corresponding positive JeKo-1 subset. Both treatment strategies were successful in sequential targeting. Either starting with LLE-CD19 mAb and in a second step by adding LLE-CD20 mAb after 48 h or vice versa. Solely, the corresponding positive tumor cell subsets were sequentially eliminated by addition of the LLE-mAb. The repetitive and sequential targeting demonstrates the serial killing capacity under various circumstances indicating the cellular functional integrity of AdCAR-T (, fig. S3B).
Figure 5. Sequential combinatorial targeting mediated by AdCAR-T
AdCAR-T function as an “AND”-gate
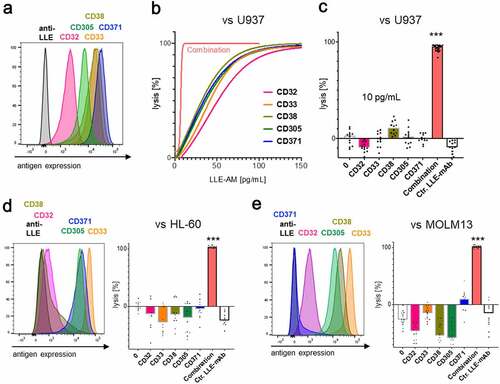
In LLE-AM titration experiments, we revealed that a certain threshold concentration of LLE-mAb bound to the target cell surface [LLE density (Tρ)] expressed on the target cell is required to activate AdCAR-T (, fig. S4A). AdCAR-T specifically bind to LLE-tags, independently of the targeted antigen (). Thus, we hypothesized that Tρ can either be reached by increasing concentrations of one single specific LLE-AM (, left panel) and likewise as the result of the assembly of LLE-AMs targeted to different antigens (Σρ = ρA+ ρB+ ρC+ … + ρX) on the target cell surface at below activation threshold concentrations (, right panel), referred to as “surface activation matrix” (SAM). To test this hypothesis, we performed titration experiments with single LLE-AMs and combinations thereof. The antigen expression of CD19 and CD20 in JeKo-1 is displayed in (). Monotargeting in JeKo-1 showed parallel lysis curves for LLE-CD19 mAb and LLE-CD20 mAb. Combining these two LLE-AMs vs. JeKo-1 at equimolar concentrations led to additive lysis (, D-E). Further, titration of monotargeting was evaluated in NALM6 for LLE-CD19 mAb, LLE-CD10 mAb, LLE-CD138 mAb, and LLE-CD22 mAb as well as the combination of these four LLE-mAbs at below threshold concentrations leading to significantly increased lysis (fig. S9A). The mechanism of additively reaching ρT (SAM) was confirmed for combining three LLE-AMs targeted to CD19, CD10, and CD138 vs. NALM6 in BCP-ALL, in neuroblastoma targeting GD2, CD81 and CD276 vs. LS and in triple negative breast cancer targeting EGFR, CD47, and CD276 vs. Hs578T (, F-H, fig. S7, A-B, D, fig. S9A). The expression level of CD32, CD33, CD38, CD305, and CD371 in the AML cell lines U937, HL-60 and MOLM13 is displayed in (, A, D-E). Encoded in the same colors, the lysis for the monotargeting is shown. To further potentiate the combinatorial effect, we combined five different AMs targeting the above listed target antigens at below threshold concentrations at the same time. Unprecedented, we found complete target elimination in combination of 5 AMs without a significant cytotoxic effect mediated by single targeting, demonstrating that AdCAR-T can function as an “AND”-gate (, B-E, fig. S7, C-D).
Figure 6. Surface Activation Matrix (SAM) a new concept of targeted polyimmunotherapy
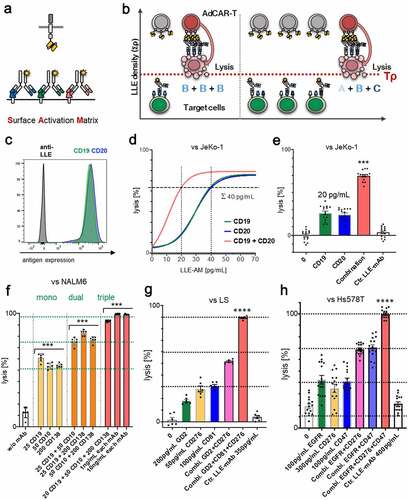
Figure 7. Identification and target cell lysis by integration of antigen expression profiles in AML
AdCAR-T selectively lyse target cells by integration of multiplex expression profiles
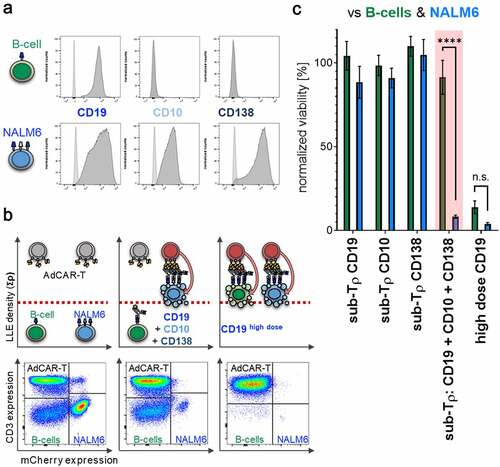
If AdCAR-T can identify target cells by multivariant antigen expression profiles, they should be capable to differentiate between cancerous and healthy tissue by rational AM selection. As a proof-of-concept, we cocultured AdCAR-T with NALM6 cells, expressing CD10, CD19, and CD138, together with freshly isolated peripheral B cells expressing solely CD19 or unmanipulated PBMCs containing B cells expressing solely CD19. Expression level of CD10, CD19 and CD138 is illustrated in (). Importantly, for B-cell positive selection CD19 REAlease® beads were used to guarantee no occupation of the CD19 extracellular domain by CD19 directed microbeads. Titration experiments showed a high intrinsic susceptibility of NALM6 to AdCAR mediated lysis and a combinatorial effect at fascinating low AM concentrations (fig. S8). Addition of an AM combination targeted to CD10, CD19, and CD138 at below threshold concentration for single targeting led to the elimination of NALM6, without significant lysis of healthy B-cells. In contrast, higher concentrations of LLE-CD19 mAb above the threshold concentration for monotargeting with LLE-CD19 mAb sufficiently eliminated both, NALM6 and healthy B cells (, B-C). Our results underscore the potential of selective cytotoxic pressure and elimination of cell subsets by AdCAR-T cells in combination with LLE-AMs at below threshold concentration based on distinct antigen expression profiles which are characteristic in malignant transformed leukemic blasts of the B lineage but also in other cancers, that do partly share potential target antigens. AdCAR-mediated cytotoxicity might be reduced by distribution and switch of AM combinations more likely to hit cancerous versus healthy tissues (fig. S10).
Figure 8. Differential target cell lysis by AdCAR-T
Discussion
CAR-T cell immunotherapy has demonstrated potent antitumor responses in B-lineage malignanciesCitation2,Citation3,Citation31,Citation32 but clinical success is limited in heterogeneous malignancies like AML and solid tumors. Consequently, there is a great medical need for controllable, flexible, and tunable CAR-T cell systems to face the challenges of acute and chronic toxicities as well as antigen escape and CAR-T exhaustion to broaden CAR-T applicability and efficacy.Citation33
Here, we developed the novel modular AdCAR-T technology for precisely controllable multiplex targeting. We demonstrate how AdCAR-T can function as a platform to address major limitations of direct CAR concepts. AdCAR-T facilitate highly specific targeting of different antigens with one single CAR construct. The AdCAR function can be tuned in and out and combinatorial targeting synchronically and sequentially counteracts antigen escape. Surface activation matrix (SAM) is introduced for the first time as a novel indirect CAR concept for differential targeted immunotherapy, and has the potential to overcome the limitations of targeting promiscuously expressed antigens by distributing on-target off-tumor toxicities. Altogether, the AdCAR-T system is a multifaceted technology for high precision adoptive CAR-T cell therapy.
Despite several modular CAR systems have been introduced,Citation14–23 none of these systems have demonstrated the same level of flexibility and versatility in generating AMs targeting a multitude of TAA (>75 AMs, data shown for 23) and utilizing different AM formats explicitly varying in size as AdCAR-T. There is a higher complexity in the optimization of newly generated AMs in peptide-based indirect CAR technologies that may complicate and slow down clinical translation,Citation20,Citation21 whereas the simplicity to achieve chemical LLE-conjugation with biotin invites to utilize any clinically available FDA/EMA-approved antibody and fragments thereof as has been demonstrated for dinutuximab, cetuximab, trastuzumab and rituximab (fig. S11).
While the structural architecture of the extracellular components in CAR T cells is crucial for effector function,Citation21,Citation26,Citation27 no impact of the AM format on effector function was observed in AdCAR-T. The AdCAR-T system allows to utilize a huge variety of different AM format (mAb, Fab2, Fab, single domain, scFv, and others) and thus combine all advantages of favorable pharmacokinetics and – dynamics, biostability and penetration into the tumor,Citation34,Citation35 adjusted to the requirements of the underlying malignancy and treatment phase (induction versus consolidation) to improve efficacy and patient safety. Moreover, convenient GMP-compliant LLE-conjugation allows economic AM generation.
In modular CAR systems, low immunogenicity of the CAR itself, the AM as well as the tag is crucial for the engraftment of the CAR-T, persistence and thus long-term functionality. Incorporation of foreign, non-human, peptide neo-epitopes or fluorochromes like FITC as tags on the AM are likely to be immunogenic.Citation20,Citation21 Leucine zippers in the zipCAR are either also non-human or in the humanized version might interfere with intracellular gene transcription and promote oncogenesis.Citation36 Human-derived peptide neo-epitopes might cause autoimmunity.Citation22 In contrast, endogenously present molecules for instance vitamins, like biotin, appear to be ideal tags. Previously described avidin- and streptavidin-based CARsCitation17,Citation18 targeted to biotin is unlikely to be translatable into the clinic, since avidin (egg-protein) as well as streptavidin (bacterial derived protein) are likely to be immunogenic.Citation37,Citation38 Potentially low immunogenicity of the AdCAR-T system will have to be demonstrated in clinical trials.
Beyond maximal flexibility in AM generation and high specificity, we address major requirements of next generation CAR design by AdCAR-T: Controllability by tunable “ON”-state (AM titration) and secure “OFF”-state in the absence of AMs. AdCAR-T can be universally redirected by exchange of the AM specificity. Consequently, AdCAR-T facilitate the treatment on a patient-individual basis utilizing off-the shelf AMs according to the individual tumor antigen expression profile. Antigen loss or downregulation has been identified in clinical studies as the major cause of treatment failure in CD19 and CD22-CAR-T therapy and is likely to play an important role in solid cancers with greater heterogeneity in antigen expressionCitation39 as shown for EGFRvIII in glioblastoma.Citation11 Substantially, we found that AdCAR-T function as an “OR”-gate by targeting of multiple antigens simultaneously. We demonstrate that trivalent targeting is capable and might prevent antigen escape by dual antigen loss. Moreover, sequential targeting underscores the flexibility to counteract antigen loss or downregulation and reallocate toxicities over the course of treatment. In vivo AdCAR-T demonstrated tumor clearance in three different B-lineage leukemia/lymphoma mouse models (NALM6, Raji, JeKo-1) utilizing two different clinical available antibodies targeted to CD19 and CD20. Benchmarking to conventional CD19CAR-T cells, considered the standard control in CAR-T cell therapy and most advanced in the clinic due to FDA approval (Kymriah® and Yescarta®), and benchmarking to conventional CD20CAR-T cells revealed slightly delayed kinetics of tumor clearance by AdCAR-T, however facilitating a higher grade of controllability, flexibility, and universal targeting.
Most remarkably, we demonstrate for the first time a technology that allows identification and differential targeting based on multivariant antigen expression profiles, “poly-AND”-gate. In contrast to previously described technologies utilizing complex T-cell engineering to enable target cell differentiation by means of recognizing only two antigens,Citation40–44 AdCAR-T differentiate target cells from normal cells by simple integration of LLE-signals presented by AMs targeting multiple different antigens at once. Our data clearly demonstrate that combinations of AMs targeting antigens expressed on the same target cell lead to AdCAR-T activation and target cell elimination at concentrations below the single activation threshold concentration of each AM. This translates in the ability to differentially lyse target cells vs. bystander cells based on multiplex partially different expression pattern (–8, fig. S7). Besides the specific SAM effect, the intrinsic susceptibility to AdCAR-mediated lysis appears to add to the differentiation of healthy versus cancerous tissue (fig. S9B). The selectivity facilitated by the SAM effect is an essential prerequisite to broaden the indications for CAR-T therapy. Since exclusive TAAs are absent and/or heterogeneously expressed in many malignancies, regulated targeting of antigens coexpressed in vitally nonessential as well as vitally essential tissues is inevitable. Applying a poly-targeting approach, including exclusively, aberrantly and/or shared expressed target antigens, the AdCAR-T system might enable to focus AdCAR-T activity on the tumor and reduce cytotoxic actions in healthy tissue coexpressing some of the targeted antigens. Overexpression as well as aberrant expression of targeted antigens then can open a therapeutic window through the SAM effect. Individual antigen-pressure could be modulated in case of intolerable on-target off-tumor activity to spare vitally essential tissues, still keeping sufficient coverage to prevent antigen escape. Further studies in relevant animal models will have to underscore the SAM-associated features of the AdCAR-T system.
In conclusion, our findings reveal insights to the unique features of the AdCAR-T technology platform that facilitates patient-individualized “poly-immunotherapy,” expected to increase efficacy by simultaneous and sequential combinatorial targeting in heterogeneous cancer entities and safety by tightly controllably function and target cell discrimination based on multiplex antigen expression profiles. The concept of AdCAR-SAM-polytargeting might facilitate CAR-T cell therapy in cancer, where no exclusive antigens are available. All components of the AdCAR-T system have been manufactured in a GMP-compliant manner allowing straightforward clinical translation. First-in-human clinical trials to demonstrate safety and efficacy are in preparation.
Materials and methods
Generation of CAR-constructs and lentiviral vectors
Transfer plasmids, based on a second-generation lentiviral vector plasmid,Citation45 were generated by cloning of the anti-LLE, anti-CD19, or anti-GD2 scFv (derived from mBio3, 4G7 and ch14.18, custom synthesized by GeneArt), on second- and third-generation CAR backbones incorporating indicated extracellular spacer and hinge domains, CD8 transmembrane domain and the cytoplasmic domains of 41BB or CD28 and 4–1BB as well as CD3ζ and truncated LNGFR (ΔLNGFR) after a F2A site. The CD20-CAR used was generated as previously described.Citation29 Lentivirus (LV) was produced in Lenti-XTM 293 T (Clontech) after lipofection (Lipofectamine 3000, Thermo Fisher) of second-generation packaging plasmid, VSV-G envelope plasmid and indicated transfer plasmid. LV containing supernatants were concentrated (Lenti-X concentrator, TaKaRa) and cryopreserved.
T-cell transduction
PBMCs were isolated from whole peripheral blood, acquired from healthy volunteer donors at the University Children’s Hospital Tuebingen, by Ficoll-Paque density gradient (Biocoll, Biochrom). T cells were isolated using anti-CD4 and anti-CD8 microbeads (Miltenyi Biotec) separately and mixed at a 1:1 ratio. T cells were activated with TransActTM (anti-CD3 and anti-CD28 agonistic signal) (Miltenyi Biotec) and cultivated in TexMACS media supplemented with 10 ng/mL IL7 and 5 ng/mL IL15 (all Miltenyi Biotec). After 24 h, activated T cells were transduced at a multiplicity of infection (MOI) of 3. Transduced T cells were maintained at 0.5–2 × 106 cells/mL in IL7/IL15 containing TexMACS media. On day 7, AdCAR transduction efficiency was determined by flow cytometry using FITC or APC conjugated mouse anti-LNGFR antibodies. In some experiments, transduced T cells were enriched using anti-LNGFR microbeads (Miltenyi Biotec). For in vivo experiments, AdCAR-Ts were generated using the TCT process on the CliniMACS ProdigyTM device as previously describedCitation29 according to the manufacturer’s instructions (Miltenyi Biotec). T cells activated with TransAct® and cultivated the same way served as control effector cells.
Adapter molecule conjugation
Adapter modification was performed at 30°C for 1 h in PBS buffer using 2–10-fold molar excess of biotin-LC-LC-NHS (Thermo Fisher Scientific, CAS-No. 89889–52–1), followed by separation of the antibody/label mix on a Sephadex G25 column. Protein containing fractions were pooled and concentration was measured by absorption at 280 nm. Successful conjugation was confirmed by LC-MS and/or by flow cytometry on cell lines expressing the target antigen and secondary staining with a fluorophore conjugated antibiotin antibody.
Generation of mAb fragments
F(ab’)2 and Fab fragments were generated by digestion of monoclonal antibodies on immobilized pepsin and papain resin (both Thermo Fisher Scientific), respectively, followed by subsequent purification on a protein A column to remove Fc fragments and undigested antibody.
Cell lines and culturing conditions
All cell lines, leukemia (NALM6, U937, MOLM-13, Kasumi-1, HL-60), lymphoma (JeKo-1, Raji) neuroblastoma (LS), and breast cancer (Hs578T) were purchased from ATCC or DSMZ (LS) and maintained in RPMI 1640 or DMEM (Hs578T) media supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Thermo Fisher Scientific), 2 mM L-glutamine and 1 mM sodium pyruvate (Biochrom), referred to as complete media. Lenti-XTM 293 T cells (Clontech) were maintained in DMEM media (Thermo Fisher Scientific) supplemented with 10% FBS, 2 mM L-glutamine and 1 mM sodium pyruvate (Biochrom). All media contained 100 units/mL of penicillin and 100 µg/mL of streptomycin (Biochrom).
Generation of luciferase expressing cell lines
Transfer plasmids, based on a third-generation lentiviral vector plasmid, containing luciferase and mCherry or GFP were kindly provided by Irmela Jeremias, Helmholtz Center Munich, Germany.Citation46 LV particles were generated as described above. Cell lines were transduced at a MOI of 3. Transgene expression was confirmed by flow cytometry using the coexpressed fluorescent protein. Transduced cells were enriched by bulk fluorescence-activated cell sorting (FACS) to maintain some degree of heterogeneity.
Generation of CD19/CD20 JeKo-1 knockout variants using CRISPR/Cas9 technology
JeKo-1 wildtype (CD19+/CD20+) was used to generate three distinct knockout variants negative for CD19 or CD20 or negative for CD19 and CD20. Electroporation was done on the Nucleofector 2b Device and the Cell Line Nucleofector Kit V (Lonza). For chemical CRISPR, 1 μg of Cas9 DNA and 1 μg of sgRNA were mixed with 2 × 106 JeKo-1 and electroporated using Nucleofector® Program M-013. After electroporation, cells were expanded in vitro. Single cell clones were generated via a depletion strategy, verified for distinct immunophenotype by flow cytometry and then used for experiments.
Quantification of cytokine release
For determination of secreted cytokines, 100.000 AdCAR-T cells per well cultivated according to the indicated conditions with added LLE-CD19 mAb plus 100.000 NALM6 cells per well in RPMI 1640-based complete media (see above) in a HERAcell incubator (Heraeus) at 37°C, 95% humidity, and 5% CO2. After 12 h, supernatants were collected.
Quantification of cytokines in the supernatants was performed via cytokine capture beads using the MACSPlex® custom cytokine assay with indicated specificities, the MACSQuant® Analyzer and the MACSQuantify® software according to the manufacturer’s instructions (Miltenyi Biotec).
Luciferase-based cytotoxicity assay (LCA)
Tumor cells were plated in RPMI 1640-based complete media (see above) at 50.000 cells per 96 flat bottom well in white plates, (Greiner bio one). Synthetic D-luciferin (Sigma Aldrich) was added at 4 µg/mL. Effector cells were plated at indicated effector to target ratio. The total volume per well was 200 µl. Therapeutic antibodies (AMs) or combinations thereof were used at indicated concentrations. Free biotin (Sigma Aldrich) and human serum (healthy donor) was used at indicated concentrations. Plates were incubated in a HERAcell incubator (Heraeus) at 37°C, 95% humidity, and 5% CO2. Plates were measured using the Wallac Victor 1420 Multilabel Counter (Perkin Elmer) at 37°C, at indicated time intervals. Lysis was determined by relative luminescence of testing condition to dilution series according to standard controls.
Real-time impedance-based cytotoxicity assay (ICA)
For label-free real-time cytotoxicity assessment, the xCELLigence device, an impedance-based Real Time Cytotoxicity Analyzer (RTCA) (ACEA Biosciences Inc.) that allows continuous assessment of cell growth and cytolysis was used.Citation47 The adherent growing neuroblastoma cell line LS was plated at 30.000 cells per well in RPMI 1640-based complete media (see above) in 96-well electronic microtiter plates E-Plate® 96 (ACEA Biosciences Inc.). After cell attachment for 24 h to achieve a robust cell index, effector cells were added according to indicated effector to target ratio. The total volume per well was 200 µl. Therapeutic antibodies (AMs) were used at indicated concentrations. Plates were incubated in a HERAcell incubator (Heraeus) under 37°C, 95% humidity, and 5% CO2 and impedance was assessed every 15 min for 24 h.
Flow cytometry-based cytotoxicity assays (antigen evasion)
To visualize antigen-specific killing, JeKo-1 wildtype, JeKo-1 CD19KO, JeKo-1 CD20 KO, and JeKo-1 CD19/CD20 KO were incubated at a 1:1:1:1 ratio with 25.000 cells per JeKo-1 variant (in total 100.000 cells) plus 200.000 AdCAR-T (E:T 2:1), according to the indicated conditions in RPMI 1640-based complete media (see above) in a HERAcell incubator (Heraeus) at 37°C, 95% humidity, and 5% CO2. Further LLE-CD19 mAb, LLE-CD20 mAb, or LLE-ROR-1 mAb and combinations thereof were added at indicated timepoints, incubated for indicated time and then analyzed on a BDTM LSR II flow cytometer. Cell populations were identified as outlined in Supplementary Figure 4b. AdCAR-T cells were identified by CD3 expression, JeKo-1 in total by GPF expression, and JeKo-1 subpopulations by CD19 and CD20 expression. Normalizing beads were used for quantification and acquisition, with a stopping gate at 100.000 beads (BDTM CompBeads per tube, Negative Control Compensation Particles, BD Biosciences). Normalized viability of target cells (JeKo-1) was calculated: Viability [%] = [(total number of target cells study condition)/(total number of target cells control condition AdCAR-T only)] × 100.
Flow cytometry-based cytotoxicity assays (differential killing)
To visualize antigen-specific differential killing, NALM6 were incubated with freshly isolated PBMCs at a 1:3 ratio with 100 000 NALM6 cells and 300.000 PBMCs plus 200.000 AdCAR-T (E:T 2:1) or freshly isolated peripheral B-cells at a 1:1 ratio with 100.000 cells each plus 200.000 AdCAR-T (E:T 2:1), according to the indicated conditions in RPMI 1640-based complete media (see above) in a HERAcell incubator (Heraeus) at 37°C, 95% humidity, and 5% CO2. Further LLE-CD19 mAb, LLE-CD10 mAb or LLE-CD138 mAb and combinations thereof were added at indicated concentration, incubated for indicated time and then analyzed on a BDTM LSR II flow cytometer. Cell populations were identified as outlined in . AdCAR-T cells were identified by CD3 expression, NALM6 by mCherry expression and B-cells by being negative for both CD3 expression and mCherry. In conditions with PBMCs, cells were stained for CD10 and CD19 to differentiate B cells and NALM6. Normalizing beads were used for quantification and acquisition, with a stopping gate at 100.000 beads (BDTM CompBeads per tube, Negative Control Compensation Particles, BD Biosciences). Peripheral B-cells were freshly isolated from PBMCs using anti-CD19 REAleaseTM microbeads (Miltenyi Biotec) according to the manufacturer’s instructions. Importantly, REAleaseTM technology facilitates the dissociation of all labels from the cell surface, ensuring that no epitopes are masked. PBMCs were isolated from whole peripheral blood, acquired from healthy volunteer donors at the University Children’s Hospital Tuebingen, by Ficoll-Paque density gradient centrifugation (Biocoll, Biochrom). Viability of target cells NALM6 and B-cells was calculated: Viability [%] = [(total number of target cells study condition)/(total number of target cells control condition AdCAR-T only)] × 100.
Antigen screening on tumor cell lines
Immunophenotyping was performed on a BDTM LSR II flow cytometer. Antibody staining was done according to standard operating procedure at 4°C in PBS buffer. Staining of tumor cells using primary labeled mAbs compared to isotype control defined antigen positivity. Further, to mimic AdCAR-T recognition, tumor cells were first stained with a primary biotinylated mAb with specificity of interest or of irrelevant specificity for isotype control. Unbound mAb was washed off carefully. Then a fluorophore labeled secondary LLE-mAb was used to semiquantify surface antigen expression. Overton positivity was calculated by integral subtraction (specific fluorescence minus fluorescence of isotype control) using FlowJo 10.4 software.
Animals and in vivo model
For all in vivo experiments 6 to 8 week old NOD.Cg-PrkdcSCID Il2rgtm1Wjl/SzJ (NSG) mice of female sex from Charles River Laboratories were used and kept in individually ventilated cages (IVC) at a maximum of five animals per cage. General health status of all animals was monitored daily. All experiments were performed according to the guidelines of the Federation of European Laboratory Animal Science Associations (FELASA) in the animal husbandry facilities of the Miltenyi Biotec GmbH in Bergisch Gladbach. On day −6 of the experiment, 0.4 × 106 Raji cells, stably expressing a firefly luciferase gene, were injected into the tail vein. Engraftment of the tumor cells was monitored by in vivo bioluminescence imaging (BLI) on day −1 and animals were randomized into groups according to tumor burden. All mice received 10 mg human IgG (Gammaguard) by intraperitoneal injection twice weekly starting on day −1. Rituximab-LLE (50 µg) was applied by intraperitoneal injection twice per week starting on day −1. CAR-T were injected intravenously on day 0. Tumor growth was monitored twice per week by BLI. Mice were sacrificed when reaching end point criteria or latest after 50 days according to the approval by the local authorities. For analysis of bone marrow, samples were taken from femur and tibia after mice were sacrificed. Cells were treated with Red Blood Cell (RBC) Lysis Buffer for 10 min and washed into PEB buffer for staining with fluorophore conjugated antibodies against huCD45-vioblue, huCD4-viogreen, huCD19-FITC, Biotin-PE, 7-AAD, huCD20-PE-Vio777, huCD271-APC, and huCD8-APC-Vio777 in order to enumerate the number of Raji lymphoma cells and AdCAR-T cells per microliter of blood. All tubes were recorded exhaustively on a MACSQuant X flow cytometer.
Optical Imaging
For tumor burden monitoring, mice were injected 3 mg D-Luciferin (Goldbio, St. Louis, MO, USA) i.p. for bioluminescence imaging (BLI) following a 5 min uptake period. An IVIS Spectrum Optical Imaging system (Perkin Elmer) was used to acquire BLI scans for 60 s or 1 s and signals were normalized for the luciferase activity [photons/second]. ROI scans were analyzed using Living Image Software 4 (Perkin Elmer), by drawing regions of interest (ROI) around the whole animal and corrected for background signal.
Stability and quantification of LLE-CD19 mAb in human whole blood and mouse plasma
For testing the stability of the LLE-conjugation to mAb and to determine the LLE-CD19 mAb concentration in human whole blood and mouse plasma we used an indirect method utilizing NALM6. In triplicates per condition 100.000 NALM6 cells were incubated at 8°C for 10 min in 50 µl of human plasma or mouse plasma containing defined concentrations of LLE-mAb (1 ng/mL, 5 ng/mL, 10 ng/mL, 25 ng/mL, 50 ng/mL, 100 ng/mL, 250 ng/mL, 500 ng/mL, and 1000 ng/mL). In a second step, a secondary antibiotin fluorophore labeled mAb (PE or APC) was used for staining after centrifugation at 500×g, 2 min in a flat bottom 96 well plate. Plate was washed with PBS twice 200 µl and after the washing step resuspended in 50 µl PBS. Secondary antibiotin mAb was added in saturation (5 µl per well) and incubation at 8°C for 10 min, twice washed and then was followed by data acquisition on a flow cytometer. The best fit line was calculated of LLE-CD19 mAb (y-axis) versus median fluorescence intensity (MFI, x-axis) and the formula was subsequently used for calculation of LLE-mAb concentrations in the linear range of the fitting line. If not in the linear range, the samples were diluted (e.g. ×2). Consequently, to determine the LLE-CD19 mAb concentration in human whole blood (plasma) or mouse plasma, NALM6 were incubated in 50 µl of the corresponding plasma and then proceeded according to the same protocol. The LLE-CD19 mAb concentration was calculated from the calculated formula. All measurements were done using at least three different plasma donors (mice or humans).
Software
Flow cytometric data was analyzed by FlowJo 10.4. Graphs and statistics were generated using the GraphPad Prism 8 software.
Statistics
All analyses were performed with GraphPad Prism 8 software. Data were presented as means ± SEM with statistically significant differences determined by tests as indicated in figure legends. P < .05 was considered as significant.
Data and Materials Availability:
All data associated with this study are present in the paper or the Supplementary Materials.
Authorship contributions:
The authors CMS, JM, SM, AK, PL, RH, and PS developed the general idea of the LLE-CAR immunotherapy project. CMS, JM, AK, and PS designed and established the AdCAR-T experiments in vitro. CMS, JM, JH, SR, CI, VK, FE, BD, GB, DA, ACK, PK, SS, NP, AR, SS, ER, ASM, AR, SS, SG, KS, DL, BK, SD, and PS performed in vitro experiments. CMS, JM, PK, MK, BP, AK, and PS designed in vivo experiments. CMS, JM, FE, BD, ACK, PK, BK, SD, and PS performed in vivo experiments. CMS, JM, SM, AK, RH, and PS interpreted the data as a whole. CMS and PS drafted and wrote the final manuscript. All authors approved the final version of this manuscript prior to submission.
Supplemental Material
Download ()Acknowledgments
The authors gratefully acknowledge Binje Vick and Irmela Jeremias for generously providing the lentiviral transfer plasmids (luciferase/mCherry and luciferase/GFP) that were used for in vitro (luciferase-based kill assays) as well as for in vivo (BLI) experiments and their expertise. We thank our collaborating Research Group Schwarzer from the Interfaculty Institute of Biochemistry (IFIB), Tübingen, Germany, especially Luisa Rehkopf and Dr. Julia Sindlinger for their technical assistance with mass spectrometry.
Disclosure statement
The employing institution of Christian M. Seitz, Jana Hau, Selina Reiter, Clara Illi, Verena Kieble, Giulia Bender, Daniel Atar, Ann-Christin Krahl, Sarah Schroeder, Nikolas Paulsen, Alexander Rokhvarguer, Sophia Scheuermann, Elena Rapp, Anna-Sophia Mast, Armin Rabsteyn, Sabine Schleicher, Stefan Grote, Karin Schilbach, Peter Lang, Rupert Handgretinger, and Patrick Schlegel (University Children’s Hospital Tuebingen) received research funding by Miltenyi Biotec GmbH on the basis of a collaboration research agreement. Furthermore, Andrew Kaiser, Joerg Mittelstaet, Stefan Miltenyi, Patrick Schlegel, Christian M. Seitz, Peter Lang, and Rupert Handgretinger are coinventors of a patent application focusing on adapter CAR technology. No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. The New England Journal of Medicine. 2017;377(26):2531–16. doi:10.1056/NEJMoa1707447.
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi:10.1056/NEJMoa1407222.
- Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. The New England Journal of Medicine. 2018;378(5):439–448. doi:10.1056/NEJMoa1709866.
- Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–195. doi:10.1182/blood-2014-05-552729.
- Mackall CL, Miklos DB. CNS Endothelial Cell Activation Emerges as a Driver of CAR T Cell-Associated Neurotoxicity. Cancer Discov. 2017;7(12):1371–1373. doi:10.1158/2159-8290.CD-17-1084.
- Gauthier J, Turtle CJ. Insights into cytokine release syndrome and neurotoxicity after CD19-specific CAR-T cell therapy. Curr Res Transl Med. 2018;66(2):50–52. doi:10.1016/j.retram.2018.03.003.
- Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015;5(12):1282–1295. doi:10.1158/2159-8290.CD-15-1020.
- Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127(20):2406–2410. doi:10.1182/blood-2015-08-665547.
- Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20–28. doi:10.1038/nm.4441.
- Shalabi H, Kraft IL, Wang HW, Yuan CM, Yates B, Delbrook C, Zimbelman JD, Giller R, Stetler-Stevenson M, Jaffe ES, et al. Sequential loss of tumor surface antigens following chimeric antigen receptor T-cell therapies in diffuse large B-cell lymphoma. Haematologica. 2018;103(5):e215–e218. doi:10.3324/haematol.2017.183459.
- O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9.
- Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350(6258):aab4077. doi:10.1126/science.aab4077.
- Sakemura R, Terakura S, Watanabe K, Julamanee J, Takagi E, Miyao K, Koyama D, Goto T, Hanajiri R, Nishida T, et al. A Tet-On Inducible System for Controlling CD19-Chimeric Antigen Receptor Expression upon Drug Administration. Cancer Immunol Res. 2016;4(8):658–668. doi:10.1158/2326-6066.CIR-16-0043.
- Cleemenceau B, Congy-Jolivet N, Gallot G, Vivien R, Gaschet J, Thibault G, Viee H. Antibody-dependent cellular cytotoxicity (ADCC) is mediated by genetically modified antigen-specific human T lymphocytes. Blood. 2006;107(12):4669–4677. doi:10.1182/blood-2005-09-3775.
- Kudo K, Imai C, Lorenzini P, Kamiya T, Kono K, Davidoff AM, et al. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. 2014;74(1):93–103. doi:10.1158/0008-5472.CAN-13-1365.
- Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, et al. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res. 2012;18(23):6436–6445. doi:10.1158/1078-0432.CCR-12-1449.
- Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, et al. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012;72(7):1844–1852. doi:10.1158/0008-5472.CAN-11-3890.
- Lohmueller JJ, Ham JD, Kvorjak M, Finn OJ. mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting. Oncoimmunology. 2017;7:e1368604.
- Kim MS, Ma JS, Yun H, Cao Y, Kim JY, Chi V, et al. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J Am Chem Soc. 2015;137(8):2832–2835. doi:10.1021/jacs.5b00106.
- Ma JS, Kim JY, Kazane SA, Choi SH, Yun HY, Kim MS, et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc Natl Acad Sci U S A. 2016;113(4):E450–458. doi:10.1073/pnas.1524193113.
- Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci U S A. 2016;113(4):E459–468. doi:10.1073/pnas.1524155113.
- Cartellieri M, Feldmann A, Koristka S, Arndt C, Loff S, Ehninger A, et al. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016;6(8):e458. doi:10.1038/bcj.2016.61.
- Cho JH, Collins JJ, Wong WW. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell. 2018;173(6):1426–1438.e11. doi:10.1016/j.cell.2018.03.038.
- Seidel UJ, Schlegel P, Grosse-Hovest L, Hofmann M, Aulwurm S, Pyz E, et al. Reduction of minimal residual disease in pediatric B-lineage acute Lymphoblastic Leukemia by an Fc-optimized CD19 Antibody. Mol Ther. 2016;24(9):1634–1643. doi:10.1038/mt.2016.141.
- Handgretinger R, Anderson K, Lang P, Dopfer R, Klingebiel T, Schrappe M, Reuland P, Gillies SD, Reisfeld RA, Niethammer D, et al. A phase I study of human/mouse chimeric antiganglioside GD2 antibody ch14.18 in patients with neuroblastoma. European Journal of Cancer. 1995;31A(2):261–267. doi:10.1016/0959-8049(94)00413-Y.
- Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015;3(2):125–135. doi:10.1158/2326-6066.CIR-14-0127.
- Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res. 2016;4(6):498–508. doi:10.1158/2326-6066.CIR-15-0231.
- Viaud S, Ma JSY, Hardy IR, Hampton EN, Benish B, Sherwood L, et al. Switchable control over in vivo CAR T expansion, B cell depletion, and induction of memory. Proc Natl Acad Sci U S A. 2018;115(46):E10898–E10906. doi:10.1073/pnas.1810060115.
- Lock D, Mockel-Tenbrinck N, Drechsel K, Barth C, Mauer D, Schaser T, et al. Automated manufacturing of potent CD20-directed chimeric antigen receptor T cells for clinical use. Hum Gene Ther. 2017;28:914–925.
- Zhu F, Shah N, Xu H, Schneider D, Orentas R, Dropulic B, et al. Closed-system manufacturing of CD19 and dual-targeted CD20/19 chimeric antigen receptor T cells using the CliniMACS Prodigy device at an academic medical center. Cytotherapy. 2018;20(3):394–406. doi:10.1016/j.jcyt.2017.09.005.
- Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-Term follow-up of CD19 CAR therapy in acute lymphoblastic Leukemia. The New England Journal of Medicine. 2018;378(5):449–459. doi:10.1056/NEJMoa1709919.
- Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33:540–549.
- Maldini CR, Ellis GI, Riley JL. CAR T cells for infection, autoimmunity and allotransplantation. Nature Reviews Immunology. 2018;18(10):605–616. doi:10.1038/s41577-018-0042-2.
- Nelson AL. Antibody fragments: hope and hype. MAbs. 2010;2(1):77–83. doi:10.4161/mabs.2.1.10786.
- Carter P. Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer. 2001;1(2):118–129. doi:10.1038/35101072.
- van Dam H, Castellazzi M. Distinct roles of Jun: Fos and Jun: ATF dimers in oncogenesis. Oncogene. 2001;20(19):2453–2464. doi:10.1038/sj.onc.1204239.
- Jain A, Barve A, Zhao Z, Jin W, Cheng K. Comparison of avidin, neutravidin, and streptavidin as nanocarriers for efficient siRNA delivery. Mol Pharm. 2017;14(5):1517–1527. doi:10.1021/acs.molpharmaceut.6b00933.
- Martorell A, Alonso E, Bone J, Echeverria L, Lopez MC, Martin F, et al. Position document: igE-mediated allergy to egg protein. Allergol Immunopathol (Madr). 2013;41(5):320–336. doi:10.1016/j.aller.2013.03.005.
- Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discovery. 2018;8(10):1219–1226. doi:10.1158/2159-8290.CD-18-0442.
- Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75.
- Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, et al. Chimeric antigen receptor t cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res. 2013;1(1):43–53. doi:10.1158/2326-6066.CIR-13-0008.
- Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, et al. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell. 2016;164(4):770–779. doi:10.1016/j.cell.2016.01.011.
- Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, et al. Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell. 2019;35(489–503):e488. doi:10.1016/j.ccell.2019.02.003.
- Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5(215):215ra172. doi:10.1126/scitranslmed.3006597.
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72(11):8463–8471. doi:10.1128/JVI.72.11.8463-8471.1998.
- Vick B, Rothenberg M, Sandhofer N, Carlet M, Finkenzeller C, Krupka C, et al. An advanced preclinical mouse model for acute myeloid leukemia using patients’ cells of various genetic subgroups and in vivo bioluminescence imaging. PLoS One. 2015;10(3):e0120925. doi:10.1371/journal.pone.0120925.
- Wing A, Fajardo CA, Posey AD Jr., Shaw C, Da T, Young RM, et al. Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus-Driven Production of a Bispecific T-cell Engager. Cancer Immunol Res. 2018;6(5):605–616. doi:10.1158/2326-6066.CIR-17-0314.