
ABSTRACT
NAD(P)H:quinone oxidoreductase 1 (NQO1) is overexpressed in most solid cancers, emerging as a promising target for tumor-selective killing. β-Lapachone (β-Lap), an NQO1 bioactivatable drug, exhibits significant antitumor effects on NQO1-positive cancer cells by inducing immunogenic cell death (ICD) and enhancing tumor immunogenicity. However, the interaction between β-Lap-mediated antitumor immune responses and neutrophils, novel antigen-presenting cells (APCs), remains unknown. This study demonstrates that β-Lap selectively kills NQO1-positive murine tumor cells by significantly increasing intracellular ROS formation and inducing DNA double strand breaks (DSBs), resulting in DNA damage. Treatment with β-Lap efficiently eradicates immunocompetent murine tumors and significantly increases the infiltration of tumor-associated neutrophils (TANs) into the tumor microenvironment (TME), which plays a crucial role in the drug’s therapeutic efficacy. Further, the presence of β-Lap-induced antigen medium leads bone marrow-derived neutrophils (BMNs) to directly kill murine tumor cells, aiding in dendritic cells (DCs) recruitment and significantly enhancing CD8+ T cell proliferation. β-Lap treatment also drives the polarization of TANs toward an antitumor N1 phenotype, characterized by elevated IFN-β expression and reduced TGF-β cytokine expression, along with increased CD95 and CD54 surface markers. β-Lap treatment also induces N1 TAN-mediated T cell cross-priming. The HMGB1/TLR4/MyD88 signaling cascade influences neutrophil infiltration into β-Lap-treated tumors. Blocking this cascade or depleting neutrophil infiltration abolishes the antigen-specific T cell response induced by β-Lap treatment. Overall, this study provides comprehensive insights into the role of tumor-infiltrating neutrophils in the β-Lap-induced antitumor activity against NQO1-positive murine tumors.
1. Introduction
Neutrophils, the most abundant subset of leukocytes in human peripheral blood, serve as the body’s first line of defense against injury, infections, and inflammation.Citation1,Citation2 Neutrophils also play a critical role in initiating acute inflammatory reactions.Citation3 Furthermore, neutrophils have emerged as significant regulators of both chronic inflammation and innate and adaptive immunity.Citation4 Like other major inflammatory cells that infiltrate tumors, neutrophils enter tumor sites under the influence of specific chemokines, cytokines, and cell adhesion molecules. However, the role of these neutrophils, known as tumor-associated neutrophils (TANs), in tumor progression remains a subject of ongoing debate.Citation5,Citation6 A growing body of evidence suggests that neutrophils can modulate the function of immune cells, exerting either suppressive or activating effects that promote tumor growth.Citation4,Citation7 Conversely, several studies highlight the antitumor effects of neutrophils, emphasizing their role in killing tumor cells, eliciting innate immune response, and inducing adaptive immunity.Citation8–11
Neutrophils isolated from blood and bone marrow have been reported to directly kill tumor cells under in vitro conditions.Citation11,Citation12 While neutrophils exhibit cytotoxic activity toward cancer cells, they can also promote tumor growth, metastasis, and immunosuppression, potentially resulting in poor patient prognosis.Citation6,Citation13,Citation14 The contrasting roles of neutrophils in tumor progression and regression may arise from their diversity and plasticity associated with maturation and activation under tumor conditions.Citation6,Citation15 The recruitment of neutrophils into the tumor microenvironment (TME) and the transition between cytotoxic, antitumor neutrophils and pro-tumorigenic neutrophils in the TME are influenced by various tumor-secreting factors, chemokines, and cytokines.Citation16–18 Furthermore, neutrophils have been found to inhibit certain types of human cancers during the early stages of tumor development or metastasis and can act as antigen-presenting cells to cytotoxic T cells.Citation11,Citation19–21 A deeper understanding of the diversity, plasticity, and transcriptional profiles of tumor-specific neutrophils has recently illuminated their antitumor properties, paving the way for the development of neutrophil-activating therapies for effective cancer treatment.Citation22–24 Moreover, recent findings suggest that neutrophils can kill antibody-opsonized cancer cells through a process termed trogoptosis, indicating that, with the right stimulation, neutrophils can elicit powerful antitumor immune responses, offering therapeutic potential against cancer.Citation21,Citation25 Chemotherapeutic drugs may alter neutrophil plasticity, resulting in diversification that could foster an immunosuppressive TME. Therefore, neutrophil-targeted therapies, such as antibody-mediated depletion, have been found to be effective. However, newer reports suggest that certain subpopulations of neutrophils can synergize with immunotherapy to enhance treatment efficacy.Citation26 So, understanding how chemotherapeutic drugs affect the plasticity and diversity of tumor-infiltrating neutrophils is important for improving cancer treatment strategies.
NAD(P)H quinone oxidoreductase 1 (NQO1) is a two-electron oxidoreductase that is overexpressed (>20–100 fold) in various solid tumors and is emerging as a valuable target for tumor-selective killing.Citation27,Citation28 NQO1 bioactivatable drugs, such as β-Lapachone (β-Lap), deoxynyboquinone, and KP372–1, activate NQO1-dependent futile oxidoreductive cycles, leading to substantial intracellular production of reactive oxygen species (ROS). These ROS have the potential to selectively induce DNA damage and cell death in tumors. This selectivity arises because normal cells possess a lower NQO1-to-catalase ratio compared to tumor cells, allowing them to efficiently scavenge ROS via catalase and thereby prevent cell death.Citation29–34 In our previous study, we found that the NQO1-targeted prodrug β-Lap induces immunogenic cell death in the tumor microenvironment (TME), triggering innate sensing and inhibiting tumor growth in a T-cell-dependent manner.Citation35 Additionally, β-Lap-mediated activation of high mobility group box 1 (HMGB1) activates the host TLR4/MyD88/type 1 interferon pathway and Batf3 dendritic cell-dependent cross-priming, bridging both innate and adaptive immune responses against the tumor.Citation35 β-Lap also demonstrated synergistic effects with anti-PD-L1 immunotherapy in overcoming adaptive resistance.Citation35 However, it remains uncertain whether the antitumor immune response mediated by β-Lap is facilitated by primary innate sensing mediators like neutrophils. The present study aims to elucidate the role of tumor-infiltrating neutrophils in the antitumor immune response following β-Lap treatment.
2. Materials and methods
2.1. Cell lines and reagents
MC38 and 4T1 cell lines were purchased from Kerafast Inc. (Boston, MA) and ATCC (Manassas, VA, USA), respectively. The EO771 cell line was generously provided by Dr. Lu at Indiana University, while the TC-1 cell line was kindly provided by Dr. T.C. Wu at Johns Hopkins University. All cell lines underwent rigorous testing for mycoplasma contamination using a mycoplasma detection kit. The cell lines were cultured in RPMI 1640 medium, or Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 1% v/v glutamine, 10% heat-inactivated fetal bovine serum, and 100 U/mL penicillin/streptomycin.
β-Lapachone (β-Lap), synthesized by Dr. Bill Bornmann (M.D. Anderson, Houston, TX), was prepared as previously described for in vitro experiments.Citation36 For in vivo studies, β-Lap was dissolved in a 20% hydroxypropyl-β-cyclodextrin (HPβCD) solution, which was prepared in 1X PBS. HPβCD (>98% purity) was purchased from Cydodextrin Technologies Development, Inc. (Gainesville, FL). Dicoumarol (DIC), Hoechst 33258, Hydrogen peroxide (H2O2), DNase I, and Histopaque 1119 and 1077 were procured from Sigma-Aldrich. OT1 peptide, Fixable viability dye eFluor 506 (eBioscience), and Gibco™ Collagenase Type IV were sourced from ThermoFisher. Anti-Ly6G (clone 1A8), anti-CD16/CD32 (clone 2.4G2), IgG (polyclonal), and anti-HMGB1 monoclonal antibodies were purchased from BioXCell (Lebanon, NH). ROS-GloTM assay kit (G8820) was obtained from Promega (Madison, WI). TACSTM Annexin V-FITC apoptosis detection kit was purchased from R&D Systems, Inc (ME, USA). IFNγ ELISPOT kit was obtained from BD Bioscience. Mouse Naïve CD8+ T cell isolation kit and PE positive selection kit were sourced from STEMCELL Technologies (Cambridge, MA). Antibodies used in this study were: γ-H2AX (JBW301, Millipore, Temecula, CA), NQO1 (A180, Santa Cruz), Tubulin (B-7, Santa Cruz), HMGB1 (3E8, company), CD45-PE/CY5 (30-F11, BioLegend, CA), CD11b-PE/CY7 (M1/70, BioLegend, CA), Ly6G-PE or Gr-1-FTIC (RV6-8C5, BioLegend, CA), Ly6C-AF488 (HK1.4, BioLegend, CA) CFSE Cell Division Tracker Kit (BioLegend, CA), CD80-PE (16-10A1, BioLegend, CA), CD86-PE/CY7 (GL-1, BioLegend, CA) CD95-APC (SA367H8, BioLegend, CA), CD54-PE (YN1/1.7.4, BioLegend, CA), TGF-β-PE (TW7-16B4, BioLegend, CA), and IFN-β-APC (Assaypro LLC, MO).
2.2. Animal acquisition and care
Female BALB/c WT and C57BL/6J WT mice, as well as female Rag1−/−, MyD88−/−, and TLR4−/− mice, all bred within the C57BL/6J genetic lineage, were purchased from The Jackson Laboratory. All mice were meticulously maintained under specific pathogen-free conditions. Animal care and experimental procedures strictly adhered to institutional and National Institutes of Health (NIH) protocols and guidelines. Ethical approval for this study was obtained from the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center and Indiana University School of Medicine.
2.3. Relative cell survival assay (DNA assay)
The relative cell survival assay was predicated on the assessment of DNA content, employing the DNA assay method as previously described.Citation36 In brief 10,000 cells were seeded into 48-well plates and subjected to varying doses of β-Lap (0–4 μM) with or without DIC (50 μM), a specific NQO1 inhibitor, for a duration of 3 h. Subsequently, the culture medium containing β-Lap was replaced with fresh medium, and the plates were incubated at 37°C for 6–7 days, allowing control cells to reach nearly 90% confluence. Following this incubation, cells were thoroughly rinsed with 1X PBS, and each well was supplemented with 250 μL of sterile distilled H2O. The plates were then subjected to overnight freezing at −80°C. On the subsequent day, cells were allowed to thaw to room temperature for cell lysis facilitation. Thereafter, 500 μL of a DNA-binding Hoechst 33258 dye solution (prepared at a final concentration of 0.01 mg/mL in 1X Tris-NaCl-EDTA buffer) was added to each well, and the plates were incubated in darkness for 2 h at room temperature. DNA content was determined by measuring the fluorescence at 460 nm using a Victor X3 plate reader (PerkinElmer Waltham, MA). The resulting data is expressed as the treated/control (T/C) ratio and is derived from hexaplicate samples (mean ± SD).
2.4. Quantification of H2O2
Murine tumor cells were cultured (1 x 104 cells/well) in 96-well white-walled clear-bottom tissue culture plates overnight and subsequently exposed to β-Lap with or without DIC for 3 h. Intracellular reactive oxygen species (ROS) induction, specifically H2O2, was quantified using the ROS-GloTM assay kit, following the manufacturer’s instructions.
2.5. Immunofluorescence staining for γ-H2AX foci
The treated murine cells were processed and stained following previously established protocols.Citation30 γ-H2AX foci, which are considered as a DNA double-strand break marker in cells, were visualized using a Leica DM5500 fluorescence microscope and quantified in terms of foci/nucleus.
2.6. Tumor growth and treatment
Approximately 6 × 105 MC38, TC-1, or EO771 cells were injected subcutaneously into the right flanks of C57BL/6 WT mice, while 6 × 105 4T1 cells were similarly injected into BALB/c WT mice. Upon tumor development, the tumor-bearing mice were randomly divided into either control or treatment groups. In the case of β-Lap treatment, tumor-bearing mice received β-Lap via intravenously at a dose of 25 mg/kg or intratumorally at dose of 18 or 22 mg/kg every other day for a total of 4 treatments. The control group was subjected to vehicle treatment, consisting of 20% HPβCD. For tumor-infiltrating neutrophil depletion, 200 μg of anti-Ly6G monoclonal antibody was administered intraperitoneally on alternate days for a total of 4 doses. For HMGB1 blockade experiments, 200 μg of anti-HMGB1 monoclonal antibody was administered intraperitoneally every 3 days for a total of 3 treatments, commencing on the same day as the initial β-lap treatment. Tumor volume was measured twice a week following the last administered dose and calculated using the following formula: (Length x Width x Width)/2.
2.7. Cell isolation from tissues
Subcutaneous tumors were harvested from mice, washed with 1X PBS, and then the tumor tissues were finely minced into small pieces. Subsequently, the tissue fragments were resuspended in a tissue digestive buffer comprising 1.5 mg/mL type 1 collagenase and 100 μg/mL DNase I, followed by a 30-minute incubation in shaking incubator at 37°C. After incubation, the tissue suspension was passed through a 70 μm cell strainer to obtain a single-cell suspension for subsequent analysis.
2.8. Flow cytometric analysis of neutrophils
The single cell suspension derived from 4T1 tumors in BALB/c WT mice, as well as TC-1, EO771, and MC38 tumors in C57BL/6 WT, MyD88−/−, TLR4−/−, or Rag−/− mice, was initially treated with anti-CD16/32 antibody (clone 2.4G2) for 10 minutes to block nonspecific binding. Subsequently, the cell suspension was incubated with fluorochrome-conjugated anti-mouse antibodies for 30 minutes at 4°C in the dark. Finally, the cell suspension was stained with Fixable viability dye eFluor 506 (eBioscience) for 10 minutes to exclude nonviable cells during flow cytometry analysis.
2.9. Co-culture of neutrophils and cancer cells
Neutrophils were isolated from the bone marrow of mice bearing 4T1 or EO771 tumors utilizing the gradient density centrifugation method, employing Histopaque 1119 and 1077.Citation37 To create antigen-induced media, 4T1 and EO771 cancer cells were cultured under various treatment conditions. For DMSO or β-Lap-induced antigen medium, cancer cells were exposed to DMSO or β-Lap (8 μM) for 4 h, followed by replacement of fresh medium for the next 48 h. Subsequently, 4T1 or EO771 cells (5 × 103) were co-cultured with bone marrow-derived neutrophils (1 × 105) for 48 h. This co-culture was conducted in the presence of complete medium (RPMI with 10% FBS) or cell culture-induced antigen, DMSO-induced antigen, or β-Lap-induced antigen medium. Cancer cell viability and death were monitored through a phase-contrast microscope and documented via photography. Cells were then incubated for 6–7 days at 37°C, and cell viability assessed by DNA assay.
2.10. Co-culture of neutrophils and CD8+ T cells
CD8+ T cells were isolated from the spleen of 4T1 tumor-bearing mice using the EasySepTM Mouse Naïve CD8+ T cell isolation kit following the manufacturer’s instructions. Tumor-associated neutrophils (TANs) were collected from 4T1 tumors (80–200 mm3) by staining the tumor cell suspension with PE-conjugated Ly6G antibody, and then the PE-tagged Ly6G+ TANs were selectively isolated using the EasySepTM PE positive selection kit as per the manufacturer’s instructions.Citation26 For CD8+ T cell proliferation analysis, purified CD8+ T cells were labeled with carboxyfluorescein diacetate N-succinimidyl ester (CFSE) and then co-cultured with the bone marrow-derived neutrophils isolated from tumor bearing mice at a 1:5 ratio for 72 h in the presence or absence of β-Lap-induced antigen. Following the 72 h co-culture, CD8+ T cell proliferation was assessed by flow cytometry, with proliferation indicated by CFSE dilution.Citation38 Subsequently, to assess T cell death induced by TANs in the presence of β-Lap, the isolated CD8+ T cells were co-cultured with TANs at a ratio of 1:5 with DMSO or β-Lap (6 μM). After 72 h of co-culture, the viability of CD8+ T cells was analyzed using the TACSTM Annexin V-FITC Apoptosis Detection Kit, with data collected on the Attune NxT Flow cytometer.
2.11. Neutrophil polarization
The polarization of bone marrow-derived neutrophils toward an antitumor N1-like phenotype was carried out in the presence or absence of β-Lap-induced antigen medium. Neutrophils (2.5 × 106 cells/mL) were isolated as previously describedCitation39 and cultured in DMSO induced antigen medium or β-Lap-induced antigen medium for 24 h. Subsequently, neutrophils were labeled for surface markers typically associated with N1-like neutrophils and then analyzed using the Attune NxT flow cytometer. Additionally, cytokines that play a role in neutrophil polarization were assessed via cytokine staining, followed by flow cytometry analysis.
2.12. Isolation of peritoneal neutrophils induced by thioglycollate broth and trans-well migration assay
Peritoneal neutrophils were isolated from tumor-bearing C57BL/6 WT or TLR4−/- mice after a 4-hour exposure to thioglycollate broth, following established protocols.Citation40 These isolated neutrophils were then utilized for migration analysis employing the trans-well technique, as previously detailed.Citation41 This migration assay involved the use of MC38 cancer cells in the presence or absence of β-Lap (4 μM), or β-Lap in combination with an anti-HMGB1 antibody.
2.13. Detection of HMGB1 release
MC38 or TC-1 cells were seeded onto a six-well plate, allowed to reach 70% confluency, and then subjected to treatment with β-Lap (2 and 4 μM), either alone or in combination with DIC (50 μM) for 3 h. Subsequently, the culture medium was replaced with fresh medium, and the supernatant was collected after 24 h. The presence of extracellular HMGB1 was assessed through western blotting.
2.14. IFNγ cytometric bead array
IFN-γ in the supernatant was quantified using a BD Cytometric Bead Array, according to the manufacturer’s protocol. After staining, samples were run on a CytoFLEX flow cytometer and data were analyzed with FlowJo software.
2.15. IFNγ enzyme-linked immunosorbent spot assay (ELISPOT)
Tumor-draining lymph nodes (TDLNs) and spleen specimens were obtained from tumor-bearing mice were collected, and single-cell suspensions were meticulously prepared. For the re-stimulation of tumor-specific T cells, irradiated tumor cells or OT-1 peptides were employed. In details, approximately 2 ~ 4 × 105 lymph node cells or splenocytes were co-cultured with 2 ~ 4 × 105 irradiated tumor cells for 48 h. Subsequently, an ELISPOT assay was performed using the IFNγ ELISPOT kit according to the manufacturer’s instructions. Spots were counted using an ImmunoSpot Analyzer (Cellular Technology Limited).
2.16. Quantitative real-time PCR
Total RNA from bone marrow-derived neutrophils (BMN) was isolated using TRIzol (Invitrogen). The quality of RNA was ensured by the 260/280 ratio using a spectrophotometer. Complementary DNA (cDNA) synthesis was carried out with the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Inc.). Quantitative real-time PCR was performed with iTaq universal SYBR Green super mix (Bio-Rad Laboratories, Inc.) according to the manufacturer’s instructions. The primers used in this study are listed in Supplementary Table 1. GAPDH was used as the housekeeping gene. The 2−ΔΔCt method was used to calculate relative expression changes.
2.17. Statistical analysis
All experimental data are expressed as mean ± SD from three independent experiments. All statistical analyses were performed with GraphPad Prism 10. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, determined by two-way ANOVA or unpaired two-tailed t-tests.
3. Results
3.1. β-Lap specifically kills NQO1+ murine tumor cells by inducing intracellular ROS generation and DNA damage
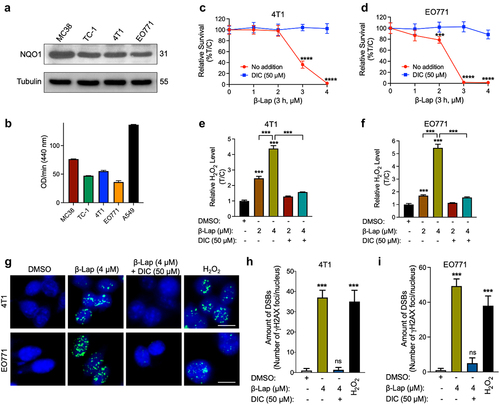
β-Lapachone (β-Lap) is a prominent NQO1 bioactivatable drugs that has shown significant antitumor effects on NQO1+ human solid cancer cell lines.Citation27,Citation29,Citation36 To assess the lethality of β-Lap in murine cell lines, we first examined the expression levels of NQO1 protein in various murine cancer cell lines through western blot analysis, as well as NQO1 enzyme activity. The results demonstrated remarkable expression of NQO1 protein in all the tested murine cancer cell lines (). Subsequently, we conducted a DNA assay to determine the cytotoxic effects of β-Lap on 4T1 and EO771 murine tumor cell lines. As expected, these NQO1+ murine cell lines exhibited sensitivity to β-Lap treatment, whereas co-treatment with dicoumarol (DIC), a specific inhibitor of NQO1, ameliorated these effects, thereby preserving cell viability (; Supplementary Fig. S1A-C). Furthermore, cytotoxicity assays conducted on MC38 WT (NQO1 over-expressing), MC38 NQO1 Knockout, B16 WT (NQO1 non-expressing), and B16 NQO1 knock-in murine tumor cells definitively demonstrated that the cell-killing efficacy of β-Lap is dependent on NQO1 expression (Supplementary Fig. S1D, E). In our previous studies, β-Lap was found to selectively kill NQO1-overexpressing human cancer cells through acute intracellular ROS generation and concomitant DNA damage.Citation29 Therefore, we investigated ROS (in the form of H2O2) generation in these murine tumor cells following β-Lap treatment and observed a significant increase in intracellular ROS levels (; Supplementary Fig. S1F, G). Additionally, we explored the effects of β-Lap treatment on DNA double strand breaks by examining Ser139-phosphorylated γH2AX foci, which serve as markers of DNA double-strand breaks (DSBs).Citation42 The results clearly indicated increased formation of γH2AX foci upon exposure to a lethal dose of β-Lap (4 μM) in 4T1 and EO771 cells, and this effect was attenuated by DIC co-treatment (). Collectively, these findings confirm that β-Lap treatment induces cell death in NQO1-positive murine tumor cells through ROS generation-associated DNA damage.
Figure 1. β-Lap selectively kills NQO1+ murine tumor cells and induces extensive DNA damage. (a–b) NQO1 expression and activity in murine tumor cell lines. (a) Level of NQO1 protein expression analyzed by western blot. (b) NQO1 enzyme activity. (c–d) Viability of murine tumor cell lines expressing endogenous NQO1; 4T1 (c) and EO771 (d) following β-Lap with or without DIC (Dicumarol, an NQO1 specific inhibitor) treatment for 3 h determined by DNA assay. (e–f) ROS measurement in murine tumor cells. (g) γH2AX foci/nuclei immunofluorescence staining indicating DNA damage in the form of double strand breaks induced by β-Lap treatment. Scale bar = 10 μm (h–i) Quantification of γH2AX foci/nuclei. Data are shown as mean ± SD from three independent experiments. ***p < 0.001, ****p < 0.0001, ns: not significant (determined by unpaired Student’s t-test).
3.2. β-Lapachone treatment leads to a significant increase in the infiltration of tumor-associated neutrophils (TANs) into the tumor microenvironment
In our previous study, it was evident that β-Lap induces immunogenic cell death (ICD) and triggers innate sensing within the tumor microenvironment (TME).Citation35 However, the specific role of β-Lap in mediating the antitumor immune response through primary mediators of innate sensing, such as neutrophils, remains unclear. To further investigate this, we examined neutrophil infiltration in the TME of different syngeneic subcutaneous mouse models, including MC38, TC-1, 4T1, and EO771 tumors. Following the inoculation of tumor cells into the mice, treatment with either a vehicle (20% HPβCD) or β-Lap was initiated when the tumor volume reached approximately 80 mm3. Treatment was administered four times on alternate days, and tumors were collected 24 hours after the final dose to prepare single-cell suspensions. Flow cytometry analysis was then performed to assess tumor-infiltrating neutrophils or tumor-associated neutrophils (TANs) using the CD11b+Gr-1high subset or the more specific CD11b+Ly6C−Ly6G+ population in vehicle- and β-Lap-treated tumors. Remarkably, we observed a significant increase in TANs infiltration in β-Lap-treated MC38, TC-1, 4T1, and EO771 tumors compared to vehicle-treated tumors, regardless of the lethal (25 mg/kg) or sublethal (18 mg/kg) dosages of β-Lap or differences in tumor cell origin (; Supplementary Fig. S2 A, B). Additionally, it was noted that MC38 tumors exhibited greater TANs infiltration among the different tumor models tested.
Figure 2. Impact of β-Lap treatment on tumor-infiltrating neutrophils in the tumor microenvironment. C57BL/6 WT mice (n = 5/group) were inoculated subcutaneously (s.c.) with 6 × 105 MC38, 1 × 106 EO771, or 1 × 105 TC-1 cells, while BALB/c WT mice (n = 5/group) received subcutaneous inoculations of 6 x 105 4T1 cells. Tumor-bearing mice were subsequently treated with β-Lap (25 mg/kg, i.v., for MC38 and TC-1, or 18 mg/kg, i.t. For 4T1 and EO771 tumors) or 20% HPβCD (Vehicle) every other day for a total of four treatment once tumor volumes reached ~80 mm3. Tumor samples were harvested 24 h after the last administrated dose. Tumor-infiltrating neutrophils were assessed in (a) MC38, (b) TC-1 [gated as CD11b+Gr-1high population], (c) 4T1 and (d) EO771 (gated with specific markers, as Ly6C−Ly6G+ population) tumors using flow cytometry. Data are presented as mean ± SD from three independent experiments. *p < 0.05, **p < 0.01 determined by unpaired Student’s t-test.
![Figure 2. Impact of β-Lap treatment on tumor-infiltrating neutrophils in the tumor microenvironment. C57BL/6 WT mice (n = 5/group) were inoculated subcutaneously (s.c.) with 6 × 105 MC38, 1 × 106 EO771, or 1 × 105 TC-1 cells, while BALB/c WT mice (n = 5/group) received subcutaneous inoculations of 6 x 105 4T1 cells. Tumor-bearing mice were subsequently treated with β-Lap (25 mg/kg, i.v., for MC38 and TC-1, or 18 mg/kg, i.t. For 4T1 and EO771 tumors) or 20% HPβCD (Vehicle) every other day for a total of four treatment once tumor volumes reached ~80 mm3. Tumor samples were harvested 24 h after the last administrated dose. Tumor-infiltrating neutrophils were assessed in (a) MC38, (b) TC-1 [gated as CD11b+Gr-1high population], (c) 4T1 and (d) EO771 (gated with specific markers, as Ly6C−Ly6G+ population) tumors using flow cytometry. Data are presented as mean ± SD from three independent experiments. *p < 0.05, **p < 0.01 determined by unpaired Student’s t-test.](/cms/asset/3aa20e9c-389f-4468-92fa-77d722d6f413/koni_a_2363000_f0002_oc.jpg)
3.3. Antibody-mediated depletion of tumor-infiltrating neutrophils abolishes the therapeutic effect of β-Lap
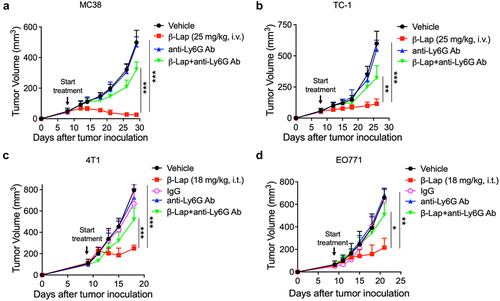
Although it is evident that there is a significant infiltration of TANs into TME following β-Lap treatment, we are curious about the impact of these TANs on the therapeutic efficacy of β-Lap. A growing body of evidence has shown that murine neutrophils can be characterized by the expression pattern of both Ly6G and Ly6C antigens, with Ly6G being specific to neutrophils.Citation43,Citation44 Furthermore, blockade of Ly6G using a neutralizing antibody (clone 1A8) treatment has been found to decrease neutrophil tissue infiltration and impair their response to injury.Citation45,Citation46 To assess the antitumor activity of β-Lap under conditions of neutrophil deficiency, we used an anti-Ly6G antibody to deplete neutrophils in C57BL/6 WT mice bearing subcutaneous MC38, TC-1 and EO771 tumors, as well as in BALB/c WT mice bearing 4T1 tumors. Mice were treated with a vehicle (20% HPβCD), β-Lap, anti-Ly6G antibody (clone 1A8), or a combination of β-Lap and anti-Ly6G antibody every other day for a total of four treatments, and tumor volume was measured to evaluate the antitumor activity. Surprisingly, depletion of tumor-infiltrating neutrophils clearly impacted on tumor regression by β-Lap at both lethal (25 mg/kg) and sub-lethal (18 mg/kg) doses. The combination treatment of anti-Ly6G antibody and β-Lap resulted in increased tumor volumes, similar to those seen with vehicle treatment, while the β-Lap alone-treated group showed a significant reduction in tumor progression across all four tumor models used in this study (). Further, flow cytometric analysis of tumor-infiltrating neutrophils (CD11b+Ly6G+ population) from anti-Ly6G antibody-treated 4T1 tumors revealed effective blockade of these neutrophils’ infiltration into the tumor (Supplementary Fig. S2C). These results indicate that β-Lap treatment suppresses tumor growth by promoting neutrophil infiltration into the TME and that these tumor-infiltrating neutrophils or TANs contribute significantly to the overall therapeutic efficacy of β-Lap.
Figure 3. Impact of neutrophil depletion on β-Lap therapeutic efficacy. (a–d) Tumor volumes measurement in MC38, TC-1, 4T1, and EO771 tumor models following neutrophil depletion. The depletion of tumor-infiltrating neutrophils was achieved using anti-Ly6G (clone 1A8) antibody, specifically targeting this neutrophil subset. IgG was employed as the anti-Ly6G control in the 4T1 and EO771 tumor models. C57BL/6 WT mice (n = 5/group) were inoculated subcutaneously (s.c.) with 6 x 105 MC38, 1 x 105 TC-1, or 1 x 106 EO771 cells, while BALB/c WT mice (n = 5/group) received subcutaneous inoculations of 6 x 105 4T1 cells. Tumor-bearing mice were subsequently treated with β-Lap (25 mg/kg, i.v., for MC38 and TC-1, or 18 mg/kg, i.t. For 4T1 and EO771 tumors), 20% HPβCD (Vehicle), anti-Ly6G (clone 1A8) antibody (200 µg, i.p.), or a combination of β-Lap+anti-Ly6G antibody every other day for a total of four times once tumor volumes reached ~80 mm3. Tumor volumes were assessed bi-weekly. Tumor volumes of MC38 (A) and TC-1 (b) in C57BL/6 mice. Tumors volumes of 4T1 in BALB/c mice (c) and EO771 in C57BL/6 mice (d). Data are shown as mean ± SD from two independent experiments. *p < 0.05, **p < 0.01 and ***p < 0.001 determined by Student’s 2-tailed t-test.
3.4. Bone marrow derived neutrophils (BMNs) not only directly kill murine tumor cells but also enhance DC recruitment and CD8+ T cell proliferation in the presence of β-Lap-induced antigen medium
To better understand the contribution of neutrophils to the antitumor activity of β-Lap, we conducted ex vivo experiments. Prior studies have reported that neutrophils can directly kill tumor cells and that high-density neutrophils (HDN) can promote CD8+ T cell proliferation in vitro.Citation11,Citation12,Citation38,Citation47 As neutrophils are produced in the bone marrow, which is considered a reliable source for their isolation,Citation37 we isolated bone marrow-derived neutrophils (BMNs) to assess their impact on tumor cell proliferation. Purity of the isolated population of bone marrow-derived high-density neutrophils was confirmed by flow cytometry (Supplementary Fig. S3A). Co-culturing BMNs with 4T1 or EO771 tumor cells for 48 h resulted in a notable reduction in tumor cell proliferation when exposed to β-Lap-induced antigen medium, compared to controls treated with cell culture- or DMSO-induced antigen medium (; Supplementary Fig. S3C, D). Furthermore, BMNs exposed to β-Lap-induced antigen medium exhibited a significant increase in reactive oxygen species (ROS) formation specifically (), whereas BMNs directly exposed to β-Lap (2 or 4 µM) for 6 or 24 hours did not elicit a similar increase in ROS (Supplementary Fig. S3E, F). These findings suggest that the activation of neutrophils is specifically mediated by the components within the β-Lap-induced antigen medium. Additionally, RT-qPCR analysis revealed an upregulation of genes associated with neutrophil activation, such as INOS and TNF-α, in BMNs cultured in β-Lap-induced antigen medium ().
Figure 4. Neutrophils kill murine tumor cells, promote DC recruitment and CD8+ T cell proliferation in the presence of β-Lap-induced antigen medium. (a–d) 4T1 or EO771 cells were exposed to cell culture medium, DMSO, or a lethal dose of β-Lap, for 4 h, followed by washing and medium replacement. After 48 h, the supernatant was collected, yielding cell culture-induced, DMSO-induced, and β-Lap-induced antigen medium, respectively. Bone marrow-derived neutrophils (BMN) were isolated through gradient density centrifugation method. Subsequently, new 4T1 or EO771 cells were co-cultured with the bone marrow-derived neutrophils in the presence or absence of respective antigens for 48 h, after which cell viability was assessed: (a) Representative image of 4T1 cells under microscope. Scale bar = 10 µm (b) Relative survival percentage of 4T1 cells in medium versus in different antigen media as determined by DNA assay. (c) Representative microscope image of EO771 cells. Scale bar = 10 µm (d) Relative survival percentage of EO771 cells determined by DNA assay. (e,f) ROS measurement from BMN culture with DMSO or β-Lap induced antigen medium for 24 h following DCFH-DA staining and flow cytometry acquisition. (g) RT-qPCR analysis for gene related immune cell recruitment (CCL5, CCL4, CCL3, CCL20) and neutrophil activation (INOS and TNF-α). (h) MC38-OVA-bearing mice (n = 3/group) were treated with β-Lap (15 mg/kg, i.t.) for one dose, and 4 days later, CD11b+Gr-1high cells were purified from the tumor drain lymph nodes, and co-cultured with CD8+ T cells isolated from the spleen of OT-1 transgenic mice. The activity of cross-priming of T cells was determined by the level of cell-secreted IFNγ via Cytometric Bead Array (CBA) mouse IFNγ assay. Naïve CD8+ T cells, isolated from the spleen of C57BL/6 OT1 mice and labeled with CFSE, were co-cultured for 3 days with bone marrow-derived neutrophils, which were either exposed to β-Lap-induced antigen medium (prepared by treating MC38-OVA cells with a lethal concentration of β-Lap or control medium: (i) Representative flow cytometry plot of OT1 CD8+ T cell proliferation. (j) Quantification of OT1 CD8+ T proliferation observed in I. Data are presented as mean ± SD from two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 determined by Student’s 2-tailed t-test.
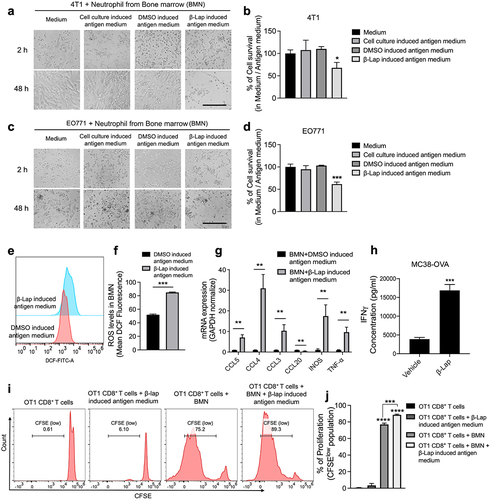
Our previous report has documented that β-Lap can induce Batf3-dependent dendritic cell-mediated T cell cross-priming.Citation35 With this in mind, we evaluated the cross-priming capability of β-Lap-induced tumor-infiltrated neutrophils using a Cytometric Bead Array (CBA) to measure IFN-γ levels secreted by T cells. The results revealed that neutrophils are also capable of cross-priming CD8+ T cells in β-Lap treated tumors, thus contributing to the antitumor immune response (). We also investigated whether neutrophils could affect DCs recruitment, maturation, or activation under β-Lap-induced antigen medium. RT-qPCR analysis showed elevated expression of chemokines such as CCL5, CCL4, and CCL3, which are essential for DC recruitment, in BMNs cultured with β-Lap-induced antigen medium compared to those cultured in DMSO-induced antigen medium (). However, no significant differences were observed in the expression of DC maturation and activation markers (CD80 and CD86) in BMNs co-cultured with immature DCs (iDCs) in the presence or absence of β-Lap-induced antigen medium (Supplementary Fig. S3G, H).
Additionally, we analyzed CD8+ T cell proliferation by co-culture of BMNs with naïve CD8+ T cells isolated from the spleens of C57BL/6J OT1 mice, with or without β-lap-induced antigen medium. Flow cytometry analysis of CFSElow OT1 CD8+ T cells, representing the proliferating CD8+ T cell population, showed a significant increase in neutrophil-CD8+ T cell co-culture with β-Lap-induced antigen medium prepared from MC38-OVA cells treated with a lethal concentration of β-lap, compared to those without β-Lap induced antigen medium (; Supplementary Fig. S4A, B). Similar tendency was also observed for OT1 CD8+ T cells co-cultured with Lipopolysaccharide (LPS, used as the model antigen) with BMNs (Supplementary Fig. S4C, D). These findings suggest β-Lap treatment promotes the proliferation of cytotoxic CD8+ T cells in the presence of neutrophils and corroborates our previous findings that β-Lap stimulates antigen-specific T cell responses in the TME.Citation35
To further investigate the impact of β-Lap-induced tumor-infiltrating neutrophils on CD8+ T cells, we sought to examine their specific effects. A previous study has highlighted the role of TANs, particularly the pro-tumorigenic N2 type, in promoting immunosuppression within the TME and inducing CD8+ T cells apoptosis.Citation48 To specifically elucidate the influence of TANs on CD8+ T cells, we co-cultured CD8+ T cells with TANs isolated from 4T1 tumors treated with β-Lap (25 mg/kg). Purity of the isolated TAN population was determined by flow cytometry (Supplementary Fig. S3B). These neutrophils were then co-cultured with naïve CD8+ T cells from spleen BALB/c mice for three days, with or without β-Lap treatment. CD8+ T cell viability was assessed using Annexin V-FITC–7AAD staining. The results demonstrated that TANs co-cultured with CD8+ T cells, with or without the lethal dose (6 μM) of β-Lap treatment, did not induce CD8+ T cell apoptosis (Supplementary Fig. S4E, F), suggesting that TANs infiltrating β-Lap-treated tumors are not detrimental to CD8+ T cells and may not belong to pro-tumorigenic N2 type.
3.5. β-Lap treatment promotes tumor associated neutrophils (TANs) polarized towards anti-tumor (N1) phenotype
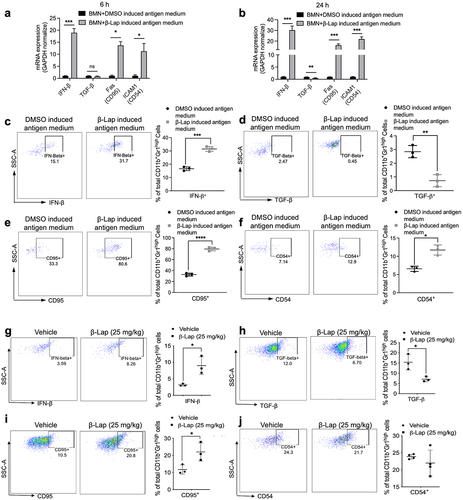
Given that our results suggest β-Lap-induced infiltrating TANs do not exhibit pro-tumorigenic N2 phenotype, we sought to determine whether these TANs exhibit antitumor N1-type characteristics. It has been reported that cytokines like IFN-β and TGF-β play roles in the polarization of neutrophils from N1 to N2 phenotype in human and murine tumors.Citation17,Citation18,Citation49 Additionally, CD95 and CD54 have been identified as typical surface markers for N1-type TANs.Citation50,Citation51 We hypothesized that β-Lap treatment induces N1 phenotypic characteristics in neutrophils. To test this, we cultured BMNs in the presence or absence of β-Lap-induced antigen medium for 24 h and assessed the gene expression of IFN-β, TGF-β, CD95, and CD54. We also measured the production of IFN-β and TGF-β cytokines, and the expression of CD95 and CD54 using flow cytometry. RT-qPCR analysis revealed a significant increase in the mRNA expression levels of IFN-β, CD95, and CD54, and a decrease in TGF-β expression in BMNs cultured in β-Lap-induced antigen medium (). Consistent with these findings, flow cytometry analysis demonstrated that neutrophils cultured in β-Lap-induced antigen medium showed significantly increased IFN-β expression and decreased TGF-β expression, compared to those cultured in DMSO-induced antigen medium (). Additionally, there was an increased in the expression of CD95 and CD54 surface markers on neutrophils grown in the β-Lap-induced antigen medium (). Furthermore, we examined these N1-type phenotypic markers in TANs infiltrated into vehicle and β-Lap-treated 4T1 tumors. Consistently, TANs from β-Lap-treated tumors displayed significantly higher expression of IFN-β and CD95, and decreased expression of TGF-β, compared to TANs from vehicle-treated tumors (). It was also noted that CD54 expression was similar in both vehicle and β-Lap-treated tumors (). These results collectively indicate that β-Lap treatment promotes the polarization of TANs toward an antitumor N1 type.
Figure 5. β-Lap treatment induces polarization of tumor associated neutrophils (TANs) towards an anti-tumor (N1) phenotype. 4T1 cells were exposed to DMSO or a lethal dose of β-Lap, for 4 h, followed by washing and medium replacement. After 48 h, the supernatant was collected, yielding β-Lap-induced antigen medium. High density neutrophils (HDN) from bone marrow of BALB/c mice were isolated through a gradient density centrifugation method and then cultured in DMSO induced antigen medium or β-Lap-induced antigen medium for 6 h or 24 h. mRNA expression levels of IFN-β, TGF-β, Fas (CD95) and ICAM1 (CD54) for (a) 6 h and (b) 24 h. (c–f) Flow cytometry was employed to analyze the expressions of cytokines at a time point of 24 h, including IFN-β (c), TGF-β (d), and surface markers CD95 (e) and CD54 (f), which are associated with anti-tumor neutrophils (N1). (g-j) BALB/c WT mice (n = 3/group) were inoculated subcutaneously (s.c.) with 6x105 4T1 cells. Once tumor volumes reached ~80 mm3, tumor-bearing mice were subsequently treated with β-Lap (18 mg/kg, i.t.) or 20% HPβCD (Vehicle) every other day for a total of four treatments. Tumor samples were harvested 24 h after the last administrated dose. The expression of N1 neutrophils-associated markers in TANs was assessed via flow cytometry: (g) Cytokine IFN-β expression, (h) TGF-β expression, (i) Surface markers CD95 expression, and (j) Surface marker CD54 expression. Data are presented as mean ± SD from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 determined by unpaired Student’s t-test.
3.6. TLR4/MyD88 signaling deficiency or HMGB1 blockade abolishes β-Lap-induced infiltration of neutrophils into the tumor microenvironment (TME)
Based on our novel findings that β-Lap treatment shapes TAN polarization toward antitumor N1 phenotype by increasing endogenous IFN-β levels, we were further curious to investigate the molecular mechanism underlying β-Lap-induced neutrophil infiltration into the TME. In our previous study, we demonstrated that type I IFNs and TLR4/MyD88 signaling were crucial for the antitumor effects of β-Lap.Citation35 Therefore, we aimed to explore the role of TLR4/MyD88 signaling in mediating β-Lap-induced neutrophil infiltration in the TME. To investigate this, we chose specifically MC38 tumors due to the observation that neutrophil infiltration is more significant in this model compared to others. We analyzed and compared the infiltration of TANs into MC38 tumors in C57BL/6 WT mice as well as MyD88-knockout (MyD88−/−) and TLR4-knockout (TLR4−/−) C57BL/6 mice following vehicle and β-Lap treatment. The results clearly showed TANs infiltration into the TME of MyD88−/− and TLR4−/− C57BL/6 mice were not significant following β-Lap treatment compared to vehicle-treated ones, while TANs infiltration was significantly increased in tumors of C57BL/6 WT mice upon β-Lap treatment (). Furthermore, we examined TANs infiltration into tumors of Rag1 knockout (Rag1−/−) mice, which lack mature B and T lymphocytes as part of the adaptive immune system. Surprisingly, we observed a significant increase in TANs in β-Lap-treated tumors of Rag1−/− mice (). These findings indicate that the infiltration of TANs into β-Lap-treated tumors is independent of the adaptive immune system, while the MyD88/TLR4 signaling pathway plays a crucial role in this process.
Figure 6. TLR4/MyD88 signaling deficiency or HMGB1 blockade significantly decreases the β-Lap-induced tumor neutrophil infiltration. (a–c) C57BL/6 WT, MyD88−/−, TLR4−/−, or Rag1−/- mice (n = 5/group) were inoculated subcutaneously (s.c.) with 6x105 MC38 cells. Tumor-bearing mice were subsequently treated with β-Lap (25 mg/kg, i.t.) or 20% HPβCD (Vehicle) every other day for a total of four cycles once tumor volumes reached ~80 mm3. Tumor samples were harvested 24 h after the last administrated dose, and tumor-infiltrating neutrophils in the tumor microenvironment were assessed via flow cytometry: (a) Neutrophil populations in MyD88−/− background tumors, (b) Percentage of neutrophils in TLR4−/− mutant tumors, and (c) Percentage of neutrophils in Rag1−/− mutant tumors. (d) Induction of HMGB1 release in the medium after 4 h of β-Lap treatment in MC38 and TC-1 cells was assessed by western blot analysis. (e) MC38 tumor models were established in C57BL/6 WT or TLR4−/− mice, following which mice were treated as described in (a). After the last administered dose, mice were treated with thioglycollate broth for 4 hours, and tumor samples were collected. The migration of peritoneal neutrophils was analyzed in MC38 cell culture medium treated with β-Lap (4 μM) or β-Lap+anti-HMGB1. (f) MC38 tumor models were established as described in (a), and then mice were treated with β-Lap ± anti-HMGB1. Anti-HMGB1 neutralized antibody (200 μg, i.p.) was administrated every 3 days for three times during the treatment. Tumor samples were harvested 24 h after the last administrated dose of β-Lap, and tumor-infiltrating neutrophils in the tumor microenvironment were assessed using flow cytometry. Data are presented as mean ± SD from two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns: not significant determined by unpaired Student’s t-test.
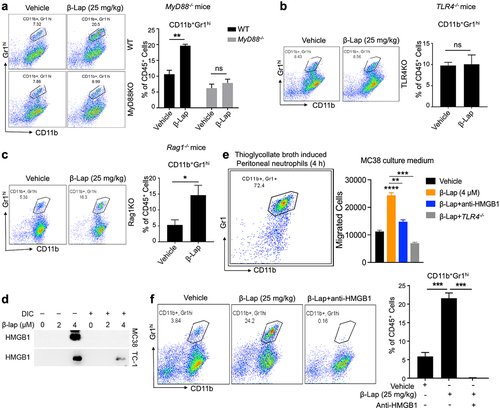
Emerging evidence suggests that high-mobility group box 1 (HMGB1) is a central damage-associated molecular pattern associated protein (DAMP) involved in neutrophil recruitment to the site of injury.Citation52 Therefore, we hypothesized that β-Lap-induced infiltration of TANs into the TME is dependent on HMGB1. To test this hypothesis, we collected culture supernatants of MC38, and TC-1 cells treated with β-Lap alone or β-Lap co-treated with DIC and performed a Western blot for HMGB1. As expected, we observed HMGB1 protein expression in samples treated with the lethal dose of β-Lap (4 µM), while co-treatment with DIC abolished the β-Lap-mediated induction of HMGB1 (). It has also been reported that the reduced form of HMGB1 is involved in leukocyte recruitment to tissue injury sites compared to its oxidized isoform.Citation53 Considering this, we also checked for the redox status of HMGB1 released into the culture supernatant of murine tumor cells treated with β-Lap by employing non-reducing SDS-PAGE and subsequent Western blot analysis. The results indicate that β-Lap treatment induces the release of reduced HMGB1, which could promote neutrophil infiltration in β-Lap-treated tumors (Supplementary Fig. S5). Next, we examined neutrophil migration toward MC38 tumor cells under ex vivo conditions using Thioglycollate broth-induced peritoneal neutrophils in a transwell assay. Interestingly, we found a significant migration of neutrophils toward β-Lap-treated MC38 cells compared to vehicle (DMSO)-treated cells. However, co-treatment of β-Lap with anti-HMGB1 antibody resulted in a significant decrease in neutrophil migration (). Additionally, neutrophils from TLR4−/− showed insignificant migration toward MC38 cells, even in the presence of β-Lap (). Furthermore, we analyzed TANs infiltration into the TME of MC38 tumors following co-treatment of β-Lap with anti-HMGB1 antibody to block HMGB1. As expected, compared to tumors treated with β-Lap alone, TANs infiltration was completely inhibited in tumors co-administrated with β-Lap and anti-HMGB1 antibody, indicating the involvement of HMGB1 in neutrophil recruitment into the TME of β-Lap-treated tumors (). Taken together, these novel findings suggest that the HMGB1/TLR4/MyD88 signaling cascade plays a key role in β-Lap-induced TANs infiltration into the tumor microenvironment.
3.7. Depletion of neutrophils, blockade of HMGB1, or deficiency in TLR4/MyD88 signaling significantly compromises the antigen-specific T cell response induced by β-Lap treatment
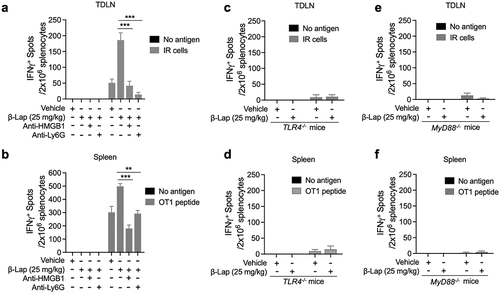
It has been demonstrated that β-Lap treatment suppresses tumor growth by enhancing tumor-specific cytotoxic CD8+ T cell response.Citation35 Furthermore, our above study indicates the role of tumor-infiltrating neutrophils in the antitumor activity of β-Lap, as well as the involvement of the HMGB1/TLR4/MyD88 signaling cascade in neutrophil infiltration into the TME. Therefore, we sought to investigate whether antibody-mediated neutrophil or HMGB1 depletion, as well as TLR4 or MyD88 knockout, affects the antigen-specific T cell response induced by β-Lap treatment in terms of the effector function of activated T cells, as indicated by IFNγ production analyzed through an ELISPOT assay. To test this hypothesis, we initially inoculated MC38 tumor cells into C57BL/6 WT mice. Once tumors were established, tumor-bearing mice were treated with β-Lap (25 mg/kg, i.v.) for alternate days for a total of three times, with or without anti-Ly6G and anti-HMGB1 antibodies to deplete TANs and HMGB1 protein, respectively. Three days after the final treatment, lymphocytes from the tumor-draining lymph nodes (TDLN) were isolated and stimulated with medium or MC38 cells irradiated with 60 Gy (for tumor antigen induction), followed by an IFNγ ELISPOT assay. Interestingly, the number of IFNγ spots was higher in the β-Lap alone treatment group, indicating an increased presence of tumor-reactive effector T cells in TDLN (). However, the population of tumor-reactive T cells was significantly reduced in TDLN when β-Lap was administered in combination with anti-Ly6G or anti-HMGB1 antibodies, as evidenced by a smaller number of IFNγ spots (). Next, we assessed IFNγ production by tumor-reactive T cells from the spleen of MC38-OVA tumor-bearing mice following the same treatment conditions. T cells isolated from the spleen were stimulated with medium or OT1 peptide, and IFNγ spots were determined. As expected, β-Lap treatment alone significantly enhanced the presence of IFNγ-producing tumor-reactive T cells compared to β-Lap treatment combined with anti-Ly6G or anti-HMGB1 antibodies (). The expansion of tumor-reactive T cells indicates that β-Lap treatment induces cross-priming and promotes T cell reactivation to suppress tumor growth. Similarly, we examined the level of tumor-reactive T cells both in TDLN and spleen from MC38 or MC38 OVA tumor-bearing TLR4−/− and MyD88−/− C57BL/6 mice treated with vehicle or β-Lap (25 mg/kg, i.v.). Remarkably, T cells isolated from TLR4−/− and MyD88−/− C57BL/6 mice exhibited significantly lower numbers of IFNγ spots, indicating a reduced level of tumor-reactive T cells compared to WT mice (). Collectively, these results suggest that neutrophil depletion, HMGB1 blockade, and TLR4/MyD88 knockout compromises the antigen-specific T cell response induced by β-Lap treatment.
Figure 7. HMGB1 blockade, neutrophils depletion, or TLR4/MyD88 deficiency significantly decreases the antigen-specific T cell response. C57BL/6 WT, TLR4−/−, or MyD88−/− mice (n = 5/group) were transplanted with 6 × 105 MC38 (a, c, e) or 1 × 106 MC38-OVA (b, d, f) cells and treated with β-Lap (25 mg/kg, i.v.) or 20% HPβCD (Vehicle) every other day for three times once tumor volumes reached ~80 mm3. 3 days after the last treatment, lymphocytes from the spleens and tumor-draining lymph nodes (TDLN) were isolated and stimulated in vitro with medium, MC38 cells irradiated with 60 Gy, or stimulated with 2.5 μg/mL of OT1 peptide for 3 days. The production of IFNγ by these stimulated cells was quantified using an ELISPOT assay. Data are presented as mean ± SD from two independent experiments. **p < 0.01, ***p < 0.001 determined by two-way ANOVA.
4. Discussion
Building on our previous studies on the antitumor activity of the NQO1-targeting prodrug β-Lap, we have found that this drug can selectively eliminate murine cell lines in an NQO1-dependent manner by inducing intracellular ROS production and DNA damage. Prior studies have shown that NQO1 bioactivatable drugs like β-Lap, deoxynyboquinone, and KP372-1 are catalyzed by the NQO1 enzyme in the presence of NAD(P)H, leading to the generation of substantial amounts of superoxide. This superoxide is then converted into membrane-permeable H2O2, which can cause DNA damage in the form of single-strand or double-strand breaks (SSBs or DSBs). The accumulation of these DNA lesions triggers the hyperactivation of poly-(ADP-ribose) polymerase-1 (PARP1), which is essential for DNA repair. This process depletes the NAD+/ATP pool and ultimately culminates in Ca2+-dependent programmed necrosis of NQO1+ cancer cells. In contrast, normal cells with sufficient catalase expression can escape the lethality induced by NQO1 bioactivatable drugs.Citation27,Citation29–31,Citation54,Citation55
Recent studies have increasingly shown that anticancer agents can induce immunogenic cell death (ICD) in the tumor microenvironment, which in turn can stimulate an anti-tumor immune response against various types of cancer. Programmed necrosis is also considered an inducer of ICD, as the release of damage-associated molecular patterns (DAMPs) can activate innate and adaptive immune responses, ultimately converting an immunosuppressive tumor microenvironment (TME) into an immunogenic one.Citation56,Citation57 Earlier research on the antitumor immune response of β-Lap provided insights into how this NQO1 bioactivatable prodrug could trigger innate sensing in the TME and induce adaptive T cell responses.Citation35 Extending these findings, our new data show the therapeutic effects of β-Lap on murine tumors originating from different tissues, including the colon (MC38), lung (TC-1), and breast (4T1 and EO771). Interestingly, in this present study, we found that the antitumor activity of β-Lap also depends on the presence of tumor-infiltrating neutrophils or tumor-associated neutrophils (TANs). Blocking the infiltration or depleting these neutrophils clearly promote tumor progression and abolishes the therapeutic effect of β-Lap. Neutrophils are usually the first immune cells to arrive at inflammatory sites and initiate an innate immune response. In certain contexts, neutrophils play a significant role in antitumor immune responses.Citation1,Citation2 However, the role of neutrophils in antitumor effects remains controversial, owing to their pro-tumorigenic roles such as immunosuppression through T cell killing or promotion of metastasis via neutrophil extracellular trap formation (NETosis).Citation38,Citation58 Concurrently, it has been shown that tumor-derived soluble factors like cytokines and chemokines not only recruit neutrophils but also contribute to neutrophil diversity and plasticity, thereby promoting tumor progression.Citation6,Citation15 Given these considerations, our results indicating the antitumor activity of infiltrating neutrophils in NQO1+ tumors following β-Lap treatment warrant further scrutiny.
Contributing to our in vivo results, we discovered that under ex vivo conditions, bone marrow-derived neutrophils (BMNs) are capable of directly killing tumor cells in the presence of β-Lap-induced antigen medium, which contains various antigenic factors. The direct cytotoxic role of neutrophils has been well-documented for decades.Citation12 Our previous research has identified dendritic cells as the primary antigen-presenting cells (APCs) driving the β-Lap-induced antitumor immune response.Citation35 Additionally, there are numerous reports indicating that anti-tumor N1 neutrophils within the TME release a range of alarmins, chemokines, and cytokines, which not only facilitate DC recruitment to the TME but also play crucial roles in the maturation and activation of DCs, thereby initiating anti-tumor adaptive immunity.Citation59 Our RT-qPCR based analysis further supports the involvement of β-Lap-induced tumor-infiltrated neutrophils in DC recruitment to the tumor microenvironment through the secretion of various chemokines. Although some reports suggest that activated neutrophils might impair T cell activation and proliferation, akin to the effects of myeloid-derived suppressor cells (MDSCs),Citation38 the dynamics of neutrophil-T cell interactions are highly dependent on the activation states of both cell types.Citation47,Citation60 Our study on the interaction between neutrophils and T cells in co-culture conditions showed that neutrophils do not kill T cells and instead have a positive effect on T-cell proliferation when exposed to β-Lap-induced antigen medium. Moreover, our previous analysis demonstrated that the antitumor effect of β-Lap relies on cytotoxic CD8+ T cells.Citation35 Taken together, these findings suggest that in β-Lap-treated tumors, both tumor-infiltrating CD8+ T cells and neutrophils coexist to induce an antitumor immune response. It is also plausible neutrophils could potentially act as atypical antigen-presenting cells.
It is well established that the transition or polarization of antitumor N1 neutrophils to pro-tumor N2 neutrophils occurs in the TME under the influence of specific cytokines, such as IFN-β and TGF-β. Increased IFN-β production promotes polarization toward N1 neutrophils, while TGF-β modulates polarization toward N2 neutrophils.Citation17,Citation18 Moreover, endogenous activation of Type I IFN (IFN α and β) signaling transforms neutrophils into an antitumor and antimetastatic phenotype by enhancing neutrophil cytotoxicity and suppressing the formation of the pre-metastatic niche.Citation61 Ex vivo experiments revealed that β-Lap-induced antigen medium can induce IFN-β cytokine expression in bone marrow-derived neutrophils while significantly reducing TGF-β expression. These neutrophils also exhibited increased expression of typical surface markers for N1 TANs, namely CD95 and CD54.Citation50,Citation51 Interestingly, our current study showed that β-Lap-induced tumor-infiltrating neutrophils from the TME exhibited enhanced IFN-β production, indicating antitumor N1 phenotypic characteristics. Furthermore, these infiltrating neutrophils displayed decreased TGF-β expression and increased CD95 expression, supporting their antitumor N1 properties. There were reports suggesting that fractionated radiotherapy polarizes newly recruited neutrophils toward the antitumor N1 phenotype in the TME.Citation62,Citation63 Our findings demonstrate that, similar to fractionated radiotherapy, the NQO1 bioactivatable drug β-Lap induces tumor-selective killing by polarizing tumor-infiltrated neutrophils into the antitumor N1 type.
In our previous research, we found that the molecular mechanism underlying antitumor activity induced by β-Lap is reliant on the drug triggering immunogenic cell death (ICD) and releasing high mobility group box 1 (HMGB1) into the tumor microenvironment. This increases tumor immunogenicity and activates the innate immune response, leading to the induction of type I interferon (IFN) through a Toll-like receptor 4 (TLR4)/MyD88-dependent pathway. This, in turn, stimulates antitumor T cell adaptive immunity and suppresses tumor growth.Citation35 Our current findings are the first to elucidate the crucial role of the HMGB1/TLR4/MyD88 cascade in the recruitment of neutrophils to β-Lap-treated tumors. Blocking HMGB1, TLR4, or MyD88 significantly inhibits neutrophil infiltration into the TME. Several studies have established that HMGB1 is essential for neutrophil recruitment and that the HMGB1/TLR4 axis can promote neutrophil extracellular trap formation (NETosis), thereby facilitating tumor progression.Citation64,Citation65 Our results also confirm that HMGB1, induced by β-Lap treatment, is vital for neutrophil recruitment to the TME. Unlike other studies linking the HMGB1/TLR4/MyD88 signaling cascade to the pro-tumor activity of neutrophils, our findings suggest that β-Lap treatment can shift tumor-infiltrating neutrophil subset toward the antitumor N1 type. These neutrophils are critical for the antitumor efficacy of β-Lap. Moreover, our experiments examining the antigenic T cell response in β-Lap-induced antitumor immunity highlight the role of neutrophils in activating T cells for effector functions. T cells isolated from tumor-bearing mice treated with β-Lap and anti-Ly6G antibody, which depletes tumor-infiltrating neutrophils, displayed reduced numbers of tumor-reactive IFNγ-producing T cells. Likewise, blocking HMGB1, TLR4, or MyD88 had a similar impact on T cells. Notably, specialized subsets of TANs have been found to act as antigen-presenting cells during the early stages of lung cancer, facilitating antigen cross-presentation and eliciting responses from antitumor T cells.Citation8,Citation10 In colorectal cancers, infiltrating neutrophils have been shown to stimulate CD8+ T cell responsiveness and activate the T-cell receptor (TCR) complex, supporting the prognostic value of CD8+ T-cell infiltration.Citation66 The significance of CD8+ T-cell infiltration is amplified by the simultaneous infiltration of neutrophils into the tumor microenvironment, suggesting their combined potential effectively promote antitumor immunity.Citation66 In summary, neutrophils that infiltrate NQO1+ tumors, recruited under the influence of β-Lap-induced HMGB1 production, play a crucial role in the T-cell-mediated anti-tumor immune response.
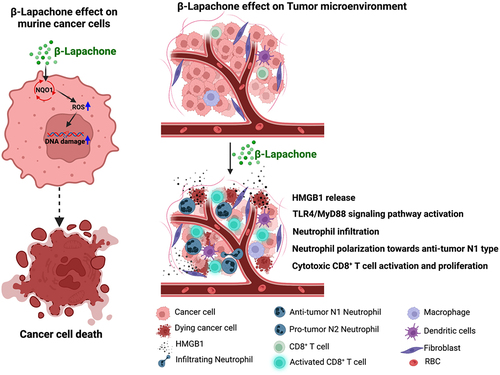
Collectively, β-Lap treatment of NQO1+ tumors induce neutrophils infiltration into the TME, and antigenic factors triggered by β-Lap polarize these neutrophils toward the antitumor N1 type. These neutrophils display direct cytotoxicity against tumor cells and contribute to innate sensing, while also coordinating with adaptive immune cells to generate antitumor immunity (). It is noteworthy that low-dose and high-dose radiation have different effects on neutrophil polarization; understanding these differences may inform synergistic therapeutic strategies to enhance radiation therapy.Citation63 Furthermore, the antitumor subpopulation of neutrophils synergizes with PD-L1-based immunotherapy in a murine lung cancer model, modulating the protumor/antitumor neutrophil ratio and enhancing the antitumor function of a specific subset of neutrophils, and promotes T cell proliferation.Citation26 Understanding the plasticity and diversity of tumor-infiltrating or tumor-associated neutrophils is beneficial for assessing therapeutic efficacy.
Figure 8. Schematic representation summarizing how β-Lap induces cell death in murine cancer cells and influences the tumor microenvironment to promote antitumor N1-type neutrophil effects (Created using BioRender.com).
In conclusion, our current study underscores the importance of the antitumor effects exerted by β-Lap-induced tumor-infiltrating neutrophils in enhancing the therapeutic efficacy of β-Lap. These findings offer promising avenues for the development of combination therapies involving β-Lap for NQO1+ tumors. Additionally, future research involving transcriptional profiling of these antitumor neutrophils could provide a more comprehensive understanding of how β-Lap influences the molecular mechanisms that underlie the antitumor properties of tumor-infiltrating neutrophils.
Abbreviations
| NQO1 | = | NAD(P)H quinone oxidoreductase-1 |
| β-Lap | = | β-Lapachone |
| ICD | = | immunogenic cell death |
| TANs | = | tumor-associated neutrophils |
| TME | = | tumor microenvironment |
| DSB | = | double-strand break |
| ROS | = | reactive oxygen species |
| DCs | = | dendritic cells |
| BMNs | = | bone marrow-derived neutrophils |
| IFN-β | = | Interferon beta |
| TGF-β | = | Transforming growth factor beta |
| HMGB1 | = | High mobility group box 1 |
| TLR4 | = | Toll Like Receptor 4 |
| MyD88 | = | Myeloid differentiation primary response 88 |
| DAMPs | = | damage-associated molecular patterns |
| NETosis | = | neutrophil extracellular trap formation |
| ATCC | = | American Tissue Culture Collection |
| RPMI 1640 | = | Roswell Park Memorial Institute 1640 |
| DMEM | = | Dulbecco’s modified Eagle’s medium |
| FBS | = | fetal bovine serum |
| DIC | = | dicoumarol |
| H2O2 | = | hydrogen peroxide |
| SSBs | = | single-strand breaks |
| RBC | = | red blood cell |
| DMSO | = | Dimethyl sulfoxide |
| ATP | = | Adenosine triphosphate |
| NAD+ | = | nicotinamide adenine dinucleotide |
Author contributions
Conceptualization: S. Tumbath, L. Jiang, X. Li and X. Huang; Methodology: S. Tumbath, L. Jiang and X. Li; Formal analysis: S. Tumbath, L. Jiang and X. Li; Investigation: S. Tumbath, L. Jiang, X. Li and T. Zhang; Statistics: S. Jiang; Writing-original draft: S. Tumbath, K. Zahid and X. Huang; Writing-review and editing: S. Tumbath, L. Jiang, Z. Yin, T. Lu, Y. Chen, X. Chen, and X. Huang; Supervision: Y. Fu and X. Huang; Resources: Y. Fu and X. Huang; Validation: Y. Fu; Project administration and Funding acquisition: X. Huang.
Highlights
β-Lap treatment significantly increases the presence of tumor-infiltrating neutrophils in the tumor microenvironment.
In the presence of antigens induced by β-Lap, bone marrow-derived neutrophils directly kill cancer cells and enhance the proliferation of CD8+ T cells.
Administration of β-Lap encourages the polarization of tumor-associated neutrophils (TANs) toward an antitumor (N1) profile and induces T cell cross-priming.
Blockade of the HMGB1/TLR4/MyD88 pathway inhibits the infiltration of neutrophils and the tumor-reactive T-cell response induced by β-Lap treatment.
Supplemental Material
Download MS Word (11.8 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Data and materials are available on reasonable request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2024.2363000.
Additional information
Funding
References
- Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol. 2014;9(1):181–17. doi:10.1146/annurev-pathol-020712-164023.
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–175. doi:10.1038/nri3399.
- Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, Catz SD. Neutrophils: New insights and open questions. Sci Immunol. 2018;3(30). doi:10.1126/sciimmunol.aat4579.
- Nicolás-Ávila JÁ, Adrover JM, Hidalgo A. Neutrophils in homeostasis, immunity, and cancer. Immunity. 2017;46(1):15–28. doi:10.1016/j.immuni.2016.12.012.
- Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis. 2012;33(5):949–955. doi:10.1093/carcin/bgs123.
- Jaillon S, Ponzetta A, Di Mitri D, Santoni A, Bonecchi R, Mantovani A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer. 2020;20(9):485–503. doi:10.1038/s41568-020-0281-y.
- Zahid KR, Raza U, Tumbath S, Jiang L, Xu W, Huang X. Neutrophils: Musketeers against immunotherapy. Front Oncol. 2022;12. doi:10.3389/fonc.2022.975981.
- Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, Stephen TL, Ranganathan A, Deshpande C, Akimova T, Vachani A, Litzky L, Hancock WW. et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J Clin Invest. 2014;124(12):5466–5480. doi:10.1172/JCI77053.
- Mensurado S, Rei M, Lança T, Ioannou M, Gonçalves-Sousa N, Kubo H, Malissen M, Papayannopoulos V, Serre K, Silva-Santos B. et al. Tumor-associated neutrophils suppress pro-tumoral IL-17+ γδ T cells through induction of oxidative stress. PLOS Biol. 2018;16(5):e2004990. doi:10.1371/journal.pbio.2004990.
- Singhal S, Bhojnagarwala PS, O’Brien S, Moon EK, Garfall AL, Rao AS, Quatromoni JG, Stephen TL, Litzky L, Deshpande C. et al. Origin and role of a subset of tumor-associated neutrophils with antigen-presenting cell features in early-stage human lung cancer. Cancer Cell. 2016;30(1):120–135. doi:10.1016/j.ccell.2016.06.001.
- Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell. 2011;20(3):300–314. doi:10.1016/j.ccr.2011.08.012.
- Clark RA, Klebanoff SJ. Neutrophil-mediated tumor cell cytotoxicity: role of the peroxidase system. J Exp Med. 1975;141(6):1442–1447. doi:10.1084/jem.141.6.1442.
- Yamanaka T, Matsumoto S, Teramukai S, Ishiwata R, Nagai Y, Fukushima M. The baseline ratio of neutrophils to lymphocytes is associated with patient prognosis in advanced gastric cancer. Oncology. 2008;73(3–4):215–220. doi:10.1159/000127412.
- Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD. et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–945. doi:10.1038/nm.3909.
- Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, Damti P, Lumbroso D, Polyansky L, Sionov RV. et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015;10(4):562–573. doi:10.1016/j.celrep.2014.12.039.
- Di Carlo E, Forni G, Lollini P, Colombo MP, Modesti A, Musiani P. The intriguing role of polymorphonuclear neutrophils in antitumor reactions. Blood. 2001;97(2):339–345. doi:10.1182/blood.V97.2.339.
- Andzinski L, Kasnitz N, Stahnke S, Wu C-F, Gereke M, von Köckritz-Blickwede M, Schilling B, Brandau S, Weiss S, Jablonska J. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int J Cancer. 2016;138(8):1982–1993. doi:10.1002/ijc.29945.
- Shaul ME, Levy L, Sun J, Mishalian I, Singhal S, Kapoor V, Horng W, Fridlender G, Albelda SM, Fridlender ZG. Tumor-associated neutrophils display a distinct N1 profile following TGFβ modulation: A transcriptomics analysis of pro- vs. antitumor TANs. Antitumor TANs, OncoImmunol. 2016;5(11):e1232221. doi:10.1080/2162402X.2016.1232221.
- Blaisdell A, Crequer A, Columbus D, Daikoku T, Mittal K, Dey SK, Erlebacher A. Neutrophils oppose uterine epithelial carcinogenesis via debridement of hypoxic tumor cells. Cancer Cell. 2015;28(6):785–799. doi:10.1016/j.ccell.2015.11.005.
- Gruijs M, Sewnath CAN, van Egmond M. Therapeutic exploitation of neutrophils to fight cancer. Semin Immunol. 2021;57:101581. doi:10.1016/j.smim.2021.101581.
- Cui C, Chakraborty K, Tang XA, Zhou G, Schoenfelt KQ, Becker KM, Hoffman A, Chang Y-F, Blank A, Reardon CA. et al. Neutrophil elastase selectively kills cancer cells and attenuates tumorigenesis. Cell. 2021;184(12):3163–3177.e3121. doi:10.1016/j.cell.2021.04.016.
- Nie P, Zhang W, Meng Y, Lin M, Guo F, Zhang H, Tong Z, Wang M, Chen F, An L. et al. AYAP/TAZ-CD54 axis is required for CXCR2−CD44− tumor-specific neutrophils to suppress gastric cancer. Protein & Cell. 2023;14(7):515–533. doi:10.1093/procel/pwac045.
- Linde IL, Prestwood TR, Qiu J, Pilarowski G, Linde MH, Zhang X, Shen L, Reticker-Flynn NE, Chiu DK, Sheu LY. et al. Neutrophil-activating therapy for the treatment of cancer. Cancer Cell. 2023;41(2):356–372.e310. doi:10.1016/j.ccell.2023.01.002.
- Matlung HL, Babes L, Zhao XW, van Houdt M, Treffers LW, van Rees DJ, Franke K, Schornagel K, Verkuijlen P, Janssen H. et al. Neutrophils kill antibody-opsonized cancer cells by trogoptosis. Cell Rep. 2018;23(13):3946–3959.e3946. doi:10.1016/j.celrep.2018.05.082.
- Zhang Y, Guoqiang L, Sun M, Lu X. Targeting and exploitation of tumor-associated neutrophils to enhance immunotherapy and drug delivery for cancer treatment. Cancer Biol Med. 2020;17(1):32–43. doi:10.20892/j.issn.2095-3941.2019.0372.
- Kaisar-Iluz N, Arpinati L, Shaul ME, Mahroum S, Qaisi M, Tidhar E, Fridlender ZG. The bilateral interplay between cancer immunotherapies and neutrophils’ phenotypes and sub-populations. Phenotypes And Sub-Popul Cells. 2022;11(5):783. doi:10.3390/cells11050783.
- Huang X, Motea EA, Moore ZR, Yao J, Dong Y, Chakrabarti G, Kilgore JA, Silvers MA, Patidar PL, Cholka A. et al. Leveraging an NQO1 bioactivatable drug for tumor-selective use of Poly(ADP-ribose) Polymerase Inhibitors. Cancer Cell. 2016;30(6):940–952. doi:10.1016/j.ccell.2016.11.006.
- Zhang K, Chen D, Ma K, Wu X, Hao H, Jiang S. NAD(P)H: Quinone oxidoreductase 1 (NQO1) as a therapeutic and diagnostic target in cancer. J Med Chem. 2018;61(16):6983–7003. doi:10.1021/acs.jmedchem.8b00124.
- Zhao W, Jiang L, Fang T, Fang F, Liu Y, Zhao Y, You Y, Zhou H, Su X, Wang J. et al. β-lapachone selectively kills hepatocellular carcinoma cells by targeting NQO1 to induce extensive DNA Damage and PARP1 Hyperactivation. Front Oncol. 2021;11:747282. doi:10.3389/fonc.2021.747282.
- Huang X, Dong Y, Bey EA, Kilgore JA, Bair JS, Li LS, Patel M, Parkinson EI, Wang Y, Williams NS. et al. An NQO1 substrate with potent antitumor activity that selectively kills by PARP1-induced programmed necrosis. Cancer Res. 2012;72(12):3038–3047. doi:10.1158/0008-5472.CAN-11-3135.
- Jiang L, Liu Y, Su X, Wang J, Zhao Y, Tumbath S, Kilgore JA, Williams NS, Chen Y, Wang X. et al. KP372-1-induced AKT hyperactivation blocks DNA repair to synergize with parp inhibitor rucaparib via Inhibiting FOXO3a/GADD45α pathway. Front Oncol. 2022;12:976292. doi:10.3389/fonc.2022.976292.
- Su X, Wang J, Jiang L, Chen Y, Lu T, Mendonca MS, Huang X. PCNA inhibition enhances the cytotoxicity of β-lapachone in NQO1-Positive cancer cells by augmentation of oxidative stress-induced DNA damage. Cancer Lett. 2021;519:304–314. doi:10.1016/j.canlet.2021.07.040.
- Chang MC, Mahar R, McLeod MA, Giacalone AG, Huang X, Boothman DA, Merritt ME. Synergistic effect of β-lapachone and aminooxyacetic acid on central metabolism in breast cancer. Nutrients. 2022;14(15):3020. doi:10.3390/nu14153020.
- Jiang L, Liu Y, Tumbath S, Boudreau MW, Chatkewitz LE, Wang J, Su X, Zahid KR, Li K, Chen Y. et al. Isopentyl-Deoxynboquinone induces mitochondrial dysfunction and g2/m phase cell cycle arrest to selectively kill NQO1-positive pancreatic cancer cells. Antioxidants & Redox Signaling. 2023; doi:10.1089/ars.2022.0224.
- Li X, Liu Z, Zhang A, Han C, Shen A, Jiang L, Boothman DA, Qiao J, Wang Y, Huang X. et al. NQO1 targeting prodrug triggers innate sensing to overcome checkpoint blockade resistance. Nat Commun. 2019;10(1):3251. doi:10.1038/s41467-019-11238-1.
- Pink JJ, Planchon SM, Tagliarino C, Varnes ME, Siegel D, Boothman DA. NAD(P)H: Quinone oxidoreductase activity is the principal determinant of β-lapachone cytotoxicity *. J Biol Chemistry. 2000;275(8):5416–5424. doi:10.1074/jbc.275.8.5416.
- Swamydas M, Luo Y, Dorf ME, Lionakis MS. Isolation of mouse neutrophils. Current Protoc Immunol. 2015;110(1):.3.20.21–.23.20.15. doi:10.1002/0471142735.im0320s110.
- Aarts CEM, Hiemstra IH, Béguin EP, Hoogendijk AJ, Bouchmal S, van Houdt M, Tool ATJ, Mul E, Jansen MH, Janssen H. et al. Activated neutrophils exert myeloid-derived suppressor cell activity damaging T cells beyond repair. Blood Adv. 2019;3(22):3562–3574. doi:10.1182/bloodadvances.2019031609.
- Sionov RV, Assi S, Gershkovitz M, Sagiv JY, Polyansky L, Mishalian I, Fridlender ZG, Granot Z. Isolation and characterization of neutrophils with anti-tumor properties. J Vis Exp. 2015;(100):e52933. doi:10.3791/52933-v.
- Luo Y, Dorf ME. Isolation of Mouse Neutrophils. Current Protoc Immunol. 1997;22(1):.3.20.21–.23.20.26. doi:10.1002/0471142735.im0320s22.
- Chan EC, Ren C, Xie Z, Jude J, Barker T, Koziol-White CA, Ma M, Panettieri RA Jr., Wu D, Rosenberg HF. et al. Regulator of G protein signaling 5 restricts neutrophil chemotaxis and trafficking. J Biol Chem. 2018;293(33):12690–12702. doi:10.1074/jbc.RA118.002404.
- Podhorecka M, Skladanowski A, Bozko P. H2AX phosphorylation: its role in DNA damage response and cancer therapy. J Nucleic Acids. 2010;2010:1–9. doi:10.4061/2010/920161.
- Lee PY, Wang JX, Parisini E, Dascher CC, Nigrovic PA. Ly6 family proteins in neutrophil biology. J Leukoc Biol. 2013;94(4):585–594. doi:10.1189/jlb.0113014.
- Hasenberg A, Hasenberg M, Männ L, Neumann F, Borkenstein L, Stecher M, Kraus A, Engel DR, Klingberg A, Seddigh P. et al. Catchup: a mouse model for imaging-based tracking and modulation of neutrophil granulocytes. Nat Methods. 2015;12(5):445–452. doi:10.1038/nmeth.3322.
- Mohamad Zaki NH, Shiota J, Calder AN, Keeley TM, Allen BL, Nakao K, Samuelson LC, Razumilava N. C-X-C motif chemokine ligand 1 induced by Hedgehog signaling promotes mouse extrahepatic bile duct repair after acute injury. Hepatology. 2022;76(4):936–950. doi:10.1002/hep.32492.
- Olofsen PA, Stip MC, Jansen JHM, Chan C, Nederend M, Tieland RG, Tsioumpekou M, Leusen JHW, Effective L-T. Neutrophil depletion using a murinized anti-ly-6G 1A8 antibody. Cells. 2022;11(21):3406. doi:10.3390/cells11213406.
- Oberg H-H, Wesch D, Kalyan S, Kabelitz D. Regulatory Interactions between neutrophils, tumor cells and T cells. Front Immunol. 2019;10. doi:10.3389/fimmu.2019.01690.
- Michaeli J, Shaul ME, Mishalian I, Hovav A-H, Levy L, Zolotriov L, Granot Z, Fridlender ZG. Tumor-associated neutrophils induce apoptosis of non-activated CD8 T-cells in a TNFα and NO-dependent mechanism, promoting a tumor-supportive environment. OncoImmunology. 2017;6(11):e1356965. doi:10.1080/2162402X.2017.1356965.
- Giese MA, Hind LE, Huttenlocher A. Neutrophil plasticity in the tumor microenvironment. Blood. 2019;133(20):2159–2167. doi:10.1182/blood-2018-11-844548.
- Wang Y, Zhao Q, Zhao B, Zheng Y, Zhuang Q, Liao N, Wang P, Cai Z, Zhang D, Zeng Y. et al. Remodeling tumor-associated neutrophils to enhance dendritic cell-based hcc neoantigen nano-vaccine efficiency. Adv Sci. 2022;9(11):2105631. doi:10.1002/advs.202105631.
- Ohms M, Möller S, Laskay T. An Attempt to Polarize Human Neutrophils Toward N1 and N2 Phenotypes in vitro. Front Immunol. 2020;11:532. doi:10.3389/fimmu.2020.00532.
- Huebener P, Pradere JP, Hernandez C, Gwak GY, Caviglia JM, Mu X, Loike JD, Schwabe RF, Antoine DJ, Schwabe RF. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest. 2015;125(2):539–550. doi:10.1172/JCI76887.
- Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, Marchis FD, Liu J, Antonelli A, Preti A, Raeli L. et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209(9):1519–1528. doi:10.1084/jem.20120189.
- Beg MS, Huang X, Silvers MA, Gerber DE, Bolluyt J, Sarode V, Fattah F, Deberardinis RJ, Merritt ME, Xie X-J. et al. Using a novel NQO1 bioactivatable drug, beta-lapachone (ARQ761), to enhance chemotherapeutic effects by metabolic modulation in pancreatic cancer. J Surg Oncol. 2017;116(1):83–88. doi:10.1002/jso.24624.
- Gerber DE, Beg MS, Fattah F, Frankel AE, Fatunde O, Arriaga Y, Dowell JE, Bisen A, Leff RD, Meek CC. et al. Phase 1 study of ARQ 761, a β-lapachone analogue that promotes NQO1-mediated programmed cancer cell necrosis. Br J Cancer. 2018;119(8):928–936. doi:10.1038/s41416-018-0278-4.
- Rapoport BL, Anderson R. Realizing the Clinical Potential of Immunogenic cell death in cancer chemotherapy and radiotherapy. Int J Mol Sci. 2019;20(4):959. doi:10.3390/ijms20040959.
- Gamrekelashvili J, Greten TF, Korangy F. Immunogenicity of necrotic cell death. Cell Mol Life Sci. 2015;72(2):273–283. doi:10.1007/s00018-014-1741-x.
- Kaltenmeier C, Simmons RL, Tohme S, Yazdani HO. Neutrophil extracellular traps (NETs) in cancer metastasis. Cancers Basel. 2021;13(23):6131. doi:10.3390/cancers13236131.
- Hadjigol S, Shah BA, O’Brien-Simpson NM. The ‘danse macabre’—neutrophils the interactive partner affecting oral cancer outcomes. Front Immunol. 2022;13. doi:10.3389/fimmu.2022.894021.
- Minns D, Smith KJ, Hardisty G, Rossi AG, Gwyer Findlay E. The outcome of neutrophil-t cell contact differs depending on activation status of both cell types. Front Immunol. 2021;12. doi:10.3389/fimmu.2021.633486.
- Pylaeva E, Lang S, Jablonska J. The essential role of type i interferons in differentiation and activation of tumor-associated neutrophils. Front Immunol. 2016;7. doi:10.3389/fimmu.2016.00629.
- Liu Q, Hao Y, Du R, Hu D, Xie J, Zhang J, Deng G, Liang N, Tian T, Käsmann L. et al. Radiotherapy programs neutrophils to an antitumor phenotype by inducing mesenchymal-epithelial transition. Transl Lung Cancer Res. 2021;10(3):1424–1443. doi:10.21037/tlcr-21-152.
- Hu J, Pan M, Wang Y, Zhu Y, Wang M. Functional plasticity of neutrophils after low- or high-dose irradiation in cancer treatment – a mini review. Front Immunol. 2023;14. doi:10.3389/fimmu.2023.1169670.
- Xiong S, Dong L, Cheng L. Neutrophils in cancer carcinogenesis and metastasis. J Hematol Oncol. 2021;14(1):173. doi:10.1186/s13045-021-01187-y.
- Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, Ballantyne CM, Gahmberg CG, Bianchi ME, Nawroth PP. et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. Embo J. 2007;26(4):1129–1139. doi:10.1038/sj.emboj.7601552.
- Governa V, Trella E, Mele V, Tornillo L, Amicarella F, Cremonesi E, Muraro MG, Xu H, Droeser R, Däster SR. et al. The interplay between neutrophils and CD8(+) T cells improves survival in human colorectal cancer. Clin Cancer Res. 2017;23(14):3847–3858. doi:10.1158/1078-0432.CCR-16-2047.