
ABSTRACT
Biomarkers for cancer immunotherapy are an unmet medical need. The group of Daniela Thommen at the NKI recently reported on novel methodologies based on short-term cultures of patient-derived tumor fragments whose cytokine concentrations in the supernatants and activation markers on infiltrating T cells were associated with clinical response to PD-1 blockade. We set up a similar culture technology with tumor-derived fragments using mouse tumors transplanted into syngeneic immunocompetent mice to test an agonist anti-CD137 mAb and its combinations with anti-PD-1 and/or anti-TGF-β. Increases in IFNγ concentrations in the tissue culture supernatants were detected upon in-culture activation with the anti-CD137 and anti-PD-1 mAb combinations or concanavalin A as a positive control. No other cytokine from a wide array was informative of stimulation with these mAbs. Interestingly, increases in Ki67 and other activation markers were substantiated in lymphocytes from cell suspensions gathered at the end of 72 h cultures. In mice bearing bilateral tumors in which one was excised prior to in vivo anti-CD137 + anti-PD-1 treatment to perform the fragment culture evaluation, no association was found between IFNγ production from the fragments and the in vivo therapeutic outcome in the non-resected contralateral tumors. The experimental system permitted freezing and thawing of the fragments with similar functional outcomes. Using a series of patient-derived tumor fragments from excised solid malignancies, we showed IFNγ production in a fraction of the studied cases, that was conserved in frozen/thawed fragments. The small tumor fragment culture technique seems suitable to preclinically explore immunotherapy combinations.
Introduction
Cancer immunotherapy advances mainly by means of deploying treatment combinations seeking synergistic effects.Citation1,Citation2 Preclinical and early clinical exploration of such approaches often poses problems of accuracy and predictability, therefore requiring complementary sources of experimental evidence.Citation3
One of the approaches that aims to solve these problems is to mimic the interface between the tumor and the immune system in tissue culture systems.Citation4 For instance, the group of Emile Voest developed organoids in coculture with autologous lymphocytes and was able to manifest signs of T-cell activation following the addition of checkpoint inhibitors to the cocultures.Citation5,Citation6 More recently, the group of Daniela Thommen, also at the NKI, developed short-term cultures of small patient-derived tumor fragments embedded in collagen to study the cytokine secretome in the supernatants and the activation of tumor-infiltrating lymphocytes (TILs) by multicolor flow cytometry.Citation7,Citation8 The latter technology has been applied to demonstrate a potentially beneficial effect of anti-PD-1 therapy in combination with COX2 inhibitionCitation9 or IL-2 addition to anti-PD-1 plus anti-CTLA-4 agents.Citation10
CD137 agonists constitute a promising tool for clinical immunotherapy following extensive evidence of efficacy in mouse models.Citation11,Citation12 At present, novel agents that target CD137 activation in the tumor lesions, bypassing liver safety issues, are under active development.Citation12 These approaches include combinations with PD-(L)1 blockade.Citation13,Citation14 Moreover, bispecific antibodies targeting PD-L1 and CD137 are under clinical development.Citation15 Further combinations potentially involving triplets or higher-order combinationsCitation16 show evidence for exceptional synergistic efficacy against difficult-to-treat mouse models.Citation17,Citation18 Hence, ex vivo models to study combined effects are of great interest in order to prioritize combinatorial developments involving CD137 agonists.
In this paper, we report on establishing a straightforward method of small tumor fragment cultures that can be stimulated with combinations based on CD137 agonists, using mouse and patient-derived excised tumors. IFNγ production released into the tissue culture supernatants was the most reliable parameter to monitor relevant pharmacodynamic effects in the cultures.
Materials and methods
Mice
C57BL/6J and Balb/c mice were purchased from Envigo RMS Spain (Barcelona, Spain). Female mice were used at 6–9 weeks of age and maintained under specific pathogen-free conditions. Animal experiments were conducted in accordance with Spanish laws and approval was obtained from the animal experimentation committee of the University of Navarra (reference: 059–21).
Tumor cell lines
MC38 and CT26 mouse colon carcinoma cell lines were kindly gifted by Dr Karl E Hellström (University of Washington, Seattle, Washington, USA) and by Mario Colombo (IRCCS Istituto Nazionale dei Tumori, Milano, Lombardia, Italy), respectively. Cells were grown in RPMI 1640 media + GlutaMAXTM (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 50 µM 2-mercaptoethanol, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37°C with 5% CO2.
Patient-derived tumor fragments
Tumor samples from patients requiring standard-of-care surgical resection were obtained from patients who signed a written informed consent (protocol ID: INTRON 2017 version 3.2, July 2018). Samples were collected between January 2021 and January 2024 at the Clínica Universidad de Navarra. A pathologist evaluated the surgical samples upon arrival at the Pathology department and provided a portion of the specimen for research use once routine pathology examinations had been performed. Tumors were collected in DMEM high glucose with GlutaMAXTM (Life Technologies) supplemented with sodium pyruvate 1 mmol/L (Life Technologies), 1× MEM nonessential AA (Life Technologies), 2 mmol/L glutamine (Life Technologies), 1% penicillin – streptomycin (Life Technologies), and 10% FBS (Sigma) and manually cut in 1 mm3 pieces on ice.
For fresh tumor sample experiments, each tumor fragment was placed in a well from 96× flat-bottom plates and overlaid in 250 μL of culture medium (see above) to which the assigned experimental agents were added. For thawed tumor samples experiments, 8–10 tumor fragments were cryopreserved in 2-ml cryotubes at −80°C in heat-inactivated 0.2 μm filtered fetal bovine serum (Sigma) containing 10% dimethyl sulfoxide (DMSO; Sigma). Upon thawing, tumor fragments were thoroughly washed in clean culture medium serially washing them on a repeatedly rinsed 70-μm cell strainer (Falcon).
Antibodies and other chemicals
Anti-mCD137 (3H3) (BE0239, BioXcell); Anti-mPD-1 (RMP1–14) (BE0146, BioXcell); Concanavalin A (C0412, Sigma); Anti-mIL-4 (BE0045, BioXcell); Rat IgG (BE0094, BioXcell); Anti-mouse TGF-β (1D11) (BE0057); Nivolumab was produced, and quality controlled at Bristol-Myers Squibb facilities (Opdivo® - a fully anti-human PD-1 IgG4); Urelumab (BMS-663513 or anti-4-1BB antibody, Bristol-Myers Squibb); Phytohemagglutinin (PHA; L1668, Sigma-Aldrich); PMA (P1585, Sigma-Aldrich); Ionomycin (I0634, Sigma-Aldrich).
Ex vivo cultures
For mouse experiments, each tumor fragment was seeded in a 96× flat-bottom well and cultured in 250 μL of culture medium (see above) in the presence of the experimental agents or corresponding controls. Cultures were supplemented with 3H3 (anti mouse-CD137) at a final concentration of 10 μg/mL, RMP1–14 (anti mouse-PD-1) at a final concentration of 10 μg/mL, 1D11 (anti human/mouse-TGF- β) at final concentration of 10 μg/mL and/or their corresponding combinations and cultured placed at 37°C and 5% CO2 for 72 hours. Culture medium was used as a negative control. Concanavalin A (ConA) at a final concentration of 10 μg/mL was used as a positive control. We set 96-well culture plates with 8–12 PDTFs per condition.
For experiments involving embedment of tumor fragments in extracellular matrix (Matrigel + Collagen) we followed previously reported methods.Citation7,Citation8
For human experiments, each tumor fragment was seeded in a 96× flat-bottom well and cultured in 250 μL of culture medium (see above) in the presence of the experimental agents or corresponding controls. Cultures were supplemented with urelumab (anti-human CD137) at a final concentration of 10 μg/mL, nivolumab (anti-human PD-1) at a final concentration of 10 μg/mL, 1D11 (anti-human/mouse pan-TGF-β) at final concentration of 10 μg/mL and/or their corresponding combinations and cultured at 37°C with 5% CO2 for 48 hours. Culture medium was used as a negative control. Phytohemagglutinin (PHA) or Phorbol 12-myristate 13-acetate (PMA) at 0,1 μg/mL plus ionomycin at a final concentration of 1 μg/mL were used as positive controls. We used 96-well culture plates with 8–10 PDTFs per condition.
Following culture, supernatants were collected and stored at −80°C until analyses of cytokine concentrations. Cell suspensions from the cultured tumor fragments were then analyzed by flow cytometry.
In vivo experiments
Six- to eight-week-old C57BL/6J and Balb/c mice were subcutaneously injected with syngeneic MC38 (5·105 cells/mouse) or CT26 (5·105 cells/mouse) cells, respectively. Therapeutic antibodies were intraperitoneally injected according to each experimental design.Citation19–21
Plasma samples were obtained from submandibular vein punctures and collected in Eppendorf tubes containing 20 μL of sodium heparin. Plasma samples were stored at −80°C until subsequent analyses.
In bilateral tumor experiments, surgical resection of one of the tumors was performed under anesthesia (intraperitoneal administration of 40 μL of a saline solution containing 45% of ketamine [Ketamidor® 100 mg/dl – Richter pharma] and 10% of xylazine [Rompun® 20 mg/dl – Bayer]. The tumor was excised with sterile surgical material. The skin wound was closed with tissue adhesive (VetBond − 3 M). Tumor growth (digital caliper) was measured twice per week. Animals which developed clinical signs of distress or severe disease (≥20% weight loss, hunched posture, reduced mobility, fur loss, tachypnea) were sacrificed according to the protocol approved by the ethics committee.
ELISA assays
Mouse IFNγ in the culture supernatants and in plasma samples was measured with a BD OptEIA™ mouse IFN-γ (AN-18) ELISA Set following manufacturer’s instructions. Human IFNγ in the culture supernatants and plasma samples was measured with a BD OptEIA™ human IFN-γ ELISA Set following manufacturer’s instructions. Mouse TGF-β1 in the culture supernatant and plasma was measured with a Duoset® ELISA Set (R&D Systems) following manufacturer’s instructions that required samples to be activated by acidification to transform TGF-β1 from the latent to the immunoreactive form.
Multiplex cytokine assays
Mouse multiplex cytokine assessments (ProcartaPlexTM Luminex platform – ThermoFisher) were performed with a customized panel of cytokines following the manufacturer’s instructions. Supernatants of wells with the same experimental conditions were pooled for these analyses.
Flow cytometry
Tumor fragments were digested with 400 U/mL collagenase D and 50 ug/mL DNase-I (Roche) for 20 minutes at 37°C. Tissue digestion was stopped by adding 3 μL of 0.5 M EDTA (Invitrogen) to each well. Single-cell suspensions were obtained by passing samples through 70 μm cell strainers (Falcon).
FcRs in cell suspensions were blocked with 10 mg/mL of beriglobin (CSL Behring, Marburg, Germany) or TruStain FcX™ PLUS (anti-mouse CD16/32) Antibody (Biolegend), respectively, in those of human or mouse origin (10 minutes at 4 °C). Samples were surface stained with the following fluorochrome-labeled antibodies in suitable combinations: anti-mouse CD3 PerCP-eFluor™ 710 (clone:17A2) (46-0032-82,eBioscience); anti-mouse CD4 BV605 (clone: GK1.5), (100451, Biolegend); anti-mouse CD8 BV510 (clone: 53–6.7), (100752, Biolegend); anti-mouse CD11b BV650 (clone : M1/70), (101259, Biolegend); anti-mouse CD19 BV650 (clone: 6D5), (115541, Biolegend); anti-mouse CD25 BV421 (clone: PC61), (102043, Biolegend); anti-mouse CD137 (clone: 17B5), (106106, Biolegend) and anti-mouse CD45 PE/Cyanine7 (clone: 30-F11) (103114 Biolegend). True-Nuclear™ Transcription Factor Buffer Set (424401, Biolegend) was used for intracellular staining. Cells were intracellularly stained with mouse anti-Ki67 AF488 (clone: B56), (558616, BD Biosciences); FoxP3 (clone: FJK-16s), (45-5773-82, eBioscience™) and anti-human/mouse Granzyme-B AF647 (clone: 6B11), (515406, Biolegend).
Human cell suspensions that had been preincubated with 10 mg/mL of beriglobin (CSL Behring, Marburg, Germany) were surface or intracellularly stained with the following fluorochrome-labeled antibodies: anti-human CD3 PCPCy5.5 (clone: UCHT1), (300430, Biolegend); anti-human CD4 PCPCy5.5 (clone: OKT4), (317428, Biolegend); anti-human CD4 BV605 (clone: RTA-T4), (562659, Biolegend); anti-human CD8 BV510 (clone: SK1), (344732, Biolegend); anti-human CD11b BV650 (clone: ICRF44), (301336, Biolegend); CD19 BV650 (clone: HIB19), (302238, Biolegend); anti-human CD25 BV421 (clone: BC96), (302238, Biolegend) and anti-human CD45 PECy7 (clone: HI30), (304016, Biolegend); anti-human CD45 BUV395 (clone: HI30), (563792, BD Bioscience). True-Nuclear™ Transcription Factor Buffer Set (424401, Biolegend) was used for intracellular staining. Cells were intracellularly stained with anti-human Ki67 AF488 (clone: Ki-67), (350508, Biolegend); anti-human Granzyme-B PE (clone: GB11), (561142, Biolegend) and anti-human/mouse Granzyme-B AF647 (clone: 6B11), (515406, Biolegend). For cell permeabilization, the eBioscienceTM FoxP3/Transcription Factor Staining Buffer Set (Invitrogen) was used following the manufacturer’s instructions.
Cell viability for gating was determined with Zombie-NIR Dye (BioLegend). The Cytoflex-S and Cytoflex-LX cytometers (Beckman Coulter) were used for data acquisition and the CytExpert v2.5 and FlowJo softwares were used for data analysis.
Confocal microscopy assessment of CD137 internalization
To perform confocal microscopy for CD137 subcellular localization in lymphocytes infiltrating tumor fragments, anti-CD137 mAb (3H3) and irrelevant RatIgG were labeled with Alexa Fluor 647 using the (Alexa Fluor™ 647 Protein Labeling Kit, Invitrogen™) according to the manufacturer´s instructions. Tumor fragments were cultured for 4 hours in the presence of Alexa Fluor647-anti CD137 or Alexa Fluor647-RatIgG. Then, fragments were recovered, washed six times in RPMI, disaggregated as previously described and the leukocytes were isolated using a Percoll™ gradient (Cytiva; 35%). Then cells were stained in FACS buffer for 10 minutes at 4°C with anti-CD8-PE (100708, Biolegend), and Hoechst 33342 to identify and exclude dead cells (1:1000 dilution, Thermo). The samples were washed twice and plated in 2% low melting agarose on 18 flat-well micro slides (IBIDI). As a control for non-internalization, fragments were cultured for 4 hours in the presence of Concanavalin A (1 mg/mL, Sigma-Aldrich), leukocytes were isolated as above and surface-stained with 3H3-Alexa Fluor 647 along with anti-CD8-PE and Hoechst 33342 in FACS buffer for 10 min at 4°C to avoid internalization at that step.
Images were taken using an 800 LSM confocal microscope (Zeiss) and analyzed using IMARIS 9 (Oxford Instruments) by defining a mask for both CD137 signal and CD8 signal. The percentage of overlap between both masks using the surface segmentation tool was calculated by the software.
Statistical analysis
Statistical analyses were performed with the Prism 8.0.2 version software (GraphPad). Continuous unpaired variables were analyzed with the Mann–Whitney test. Survival was reported following the Kaplan–Meier method and analyzed between groups with log-rank (Mantel-Cox) tests. Correlations between continuous variables were calculated with the non-parametric Spearman test and linear regression lines were represented on correlation graphs. A two-tailed p < .05 was considered statistically significant. Tumor growth differences were analyzed by two-way ANOVA tests. When differences were statistically significant, the significance is represented with asterisks (*): * for a p value < .05, ** for a p value < .01, *** for a p value < .001, **** for a p value < .0001. Statistical differences with the corresponding positive controls are not shown in figures for reasons of simplicity. Error bars indicate the standard error of the mean (SEM) unless otherwise indicated in figure legends.
Results
Cultured mouse tumor fragments respond to combinations of anti-CD137 and anti-PD-1 mAbs releasing IFNγ into the supernatants
To develop experimental tools to study immunotherapy combinations, we started culture experiments with ≈1 mm3 tumor fragments excised from MC38 and CT26 established tumors as represented in . Single fragments per tissue culture well were seeded in culture wells without extracellular matrix and supernatants and fragments collected at 72 h. This technique permitted us to incubate a series of fragments with different agents that aimed to replicate the stimulation of tumor-residing T lymphocytes at the time of surgical excision. Such activation was monitored by studying IFNγ secretion into the tissue culture supernatants and performing multicolor flow cytometry on single-cell suspensions from pooled fragments from each tumor ().
Figure 1. Tumor-infiltrating lymphocytes in cultured tumor fragments from established mouse tumors are activated by anti-PD-1 plus anti-CD137 mAb combinations.
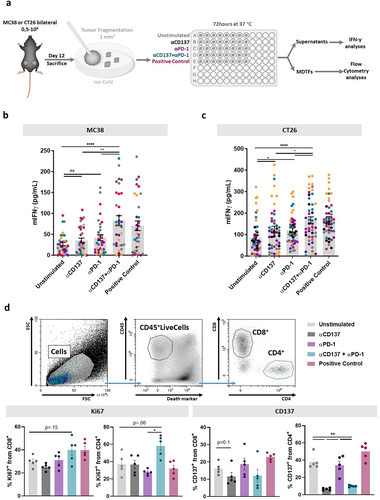
As can be seen in , the addition of anti-PD-1 and anti-CD137 mAbs in combination to the cultured tumor fragments from MC38 and CT26-derived tumors resulted in significant increases of IFNγ concentrations in the supernatants that were comparable with the positive control (concanavalin A). Furthermore, we could observe viable CD4+ and CD8+ T lymphocytes in the corresponding cell suspensions following the 72 h culture period (), that preserved the proportions of effector CD8+ and CD4+ cells with only a mild relative increase of CD4+FoxP3+ Tregs (Figure S1). In this setting, anti-CD137 and anti-PD-1 mAbs resulted in increased proportions of CD4+ and CD8+ T cells bearing the proliferation marker Ki67 ().
However, when we attempted to use CD137 as a marker of T-cell activation, we routinely observed a clear reduction of the intensity of surface staining ( and S2a). Experiments in Supplementary Figure S2 confirmed such phenomenon in CD4+ and CD8+ T cells from the tumor fragments. This observation is not attributable to epitope competition between the anti-CD137 mAbs used for stimulation and detection since they do not cross-compete.Citation22
As previously reported,Citation23 internalization was a likely cause to explain reduced surface staining. Indeed, surface CD137 staining as compared to intracellular staining supported such an explanation (Figure S2b). Moreover, when confocal microscopy was performed on tumor-infiltrating lymphocytes (TILs) from MC38 tumors exposed to the AF647-labeled CD137 agonist (3H3) mAb, clear endosomal internalization patterns were observed that could be quantified as CD137 immunostaining not colocalizing with CD8 on the cell surface (Figure S2c).
We then used similar tumor-derived cultures (Figure S3a) to determine if there were other cytokines released to the supernatants which could indicate TIL activation. Of note, only IFNγ yielded consistent results among the broad array of cytokines tested using a multiplexed ELISA panel (Figure S3b and c), that only showed minor changes in other cytokines in response to the anti-PD-1 plus anti-CD137 combination (Figure S3c).
Prior to the establishment of extracellular matrix-free cultures, we compared the influence of the embedment of fragments in extracellular matrix, as previously reported,Citation7,Citation8 to check its influence on relevant identified readouts. The addition of the extracellular matrix, as previously described,Citation7,Citation8 tended to reduce the leukocyte efflux from fragments, although a radial migration of cells was observed after 72 hours of culture also in a fraction of matrix-embedded fragments (Figure S4a). Regarding activity readouts, matrix-embedded fragments secreted less IFNγ to the supernatant in the unstimulated conditions (Figure S4b), while viability of the TILs retrieved from cultured fragments and CD137 internalization upon exposure to the agonistic antibody were similar in both culture conditions (Figure S4c-e). The effect of anti-CD137 + anti-PD1 mAbs was preserved in cultures with and without artificial extracellular matrix embedment (Figure S4b-e).
Fresh and frozen/thawed mouse tumor-derived fragments permitted culture experiments providing similar results
From the point of view of experimental logistics and experimental reproducibility, the feasibility of freezing the tissue fragments for subsequent experiments is considered an advantage. For this purpose, we froze several cryotubes of MC38- and CT26-derived tumor fragments in freezing medium that were stored at −196°C for several weeks and compared results with respect to the testing of freshly processed specimens ().
Figure 2. Tumor-derived fragments can be frozen and thawed showing comparable results with freshly excised fragments.
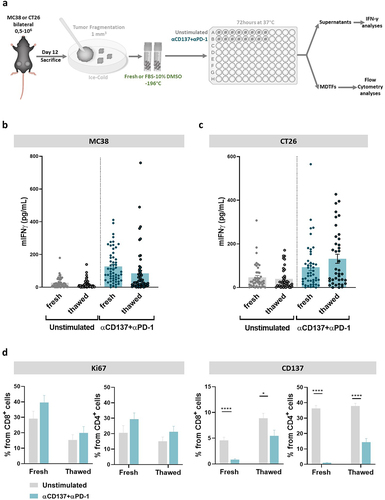
Comparisons as those shown in upon stimulation with anti-PD-1 and anti-CD137 of the fragments indicated that thawed fragments yielded a comparable release of IFNγ into the supernatants in 72 h cultures. Moreover, flow cytometry analyses on fragment-derived cell suspensions showed some level of Ki67 induction and internalization of CD137 () upon culture with the monoclonal antibody combinations under scrutiny.
Testing tumor-derived fragments to predict outcome and explore suitable additional targets to improve results
Conceivably, the maximal value of these ex-vivo experimental procedures would come from their potential to reliably predict the outcome of immunotherapy and to test novel agents and combinations. To explore the predictive capacity in mouse models, we set up experiments in mice engrafted bilaterally with MC38 tumors. In these experiments, schematically represented in , one of the tumors was surgically excised to derive fragment cultures, while the other one was left in place to monitor its outcome. Mice would subsequently receive an anti-PD-1 plus anti-CD137 or control mAb courses (). Results in show the individual follow-up of the sizes of the remaining tumor, which allowed the mice to be classified as responders or non-responders to the treatment.
Figure 3. IFNγ release into the supernatant does not predict the therapeutic response to anti-PD1 plus anti-CD137 mAb treatment in vivo.
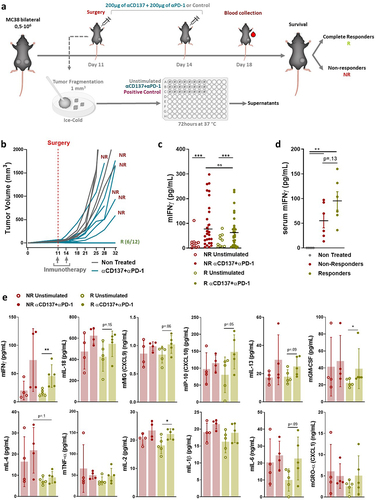
As shows, anti-CD137 plus anti-PD-1 mAb ex vivo treatment of the fragments gave similar levels of IFNγ that were measurable in the tissue culture supernatants regardless of the actual outcome of the contralateral tumor, as followed in each mouse. However, when circulating IFNγ concentrations from such mice were evaluated on day 18 of the experiment, there was a tendency to an association with the in vivo response of the tumors in each mouse ().
In these tissue culture supernatants, we sought to identify some other soluble mediators that could better predict response. The only candidate that emerged was a tendency toward higher values of interleukin-4 (IL-4) from the fragments of mice whose contralateral tumor eventually progressed (). Based on these increases of IL-4 and in light of recent studies showing that IL-4 neutralization in mice is beneficial for the outcome of checkpoint immunotherapy,Citation24 we tested whether IL-4 neutralization would add any efficacy to the anti-PD-1 plus anti-CD137 doublet. To address this point, cotreatment experiments were set up as described in Figure S5a. However, survival (Figure S5b) and tumor size follow-up (Figure S5c) indicated only a marginal benefit of the triple combination including IL-4 neutralization.
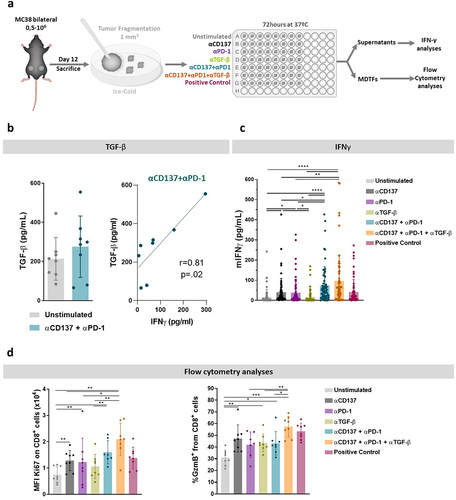
Another candidate to interfere with tumor immunity is TGF-β as extensively described in the literature.Citation25–28 Therefore, we studied whether neutralization of TGF-β1–3 would show enhanced activation of TILs in the fragments in experiments as those outlined in . Indeed, as can be seen in (left panel), fragments secreted detectable concentrations of TGF-β that were not significantly modified by anti-PD-1 plus anti-CD137 mAbs. Interestingly, the higher the IFNγ levels, the higher were the levels of TGF-β (, right panel). This suggests that TGF-β is an actionable compensatory mechanism. However, in terms of IFNγ secretion into the supernatants, we only found a tendency toward higher levels upon TGF-β co-neutralization (). Moreover, when analyzing the activation of CD8+ T cells in cell suspensions after culture of the fragments, more pronounced inductions of Ki67 and granzyme B were observed with the triplet containing TGF-β neutralization ().
Figure 4. TGF-β blockade enhances the activation of TILs in the fragments as induced by anti-CD137 in combination with anti-PD-1 plus anti-CD137 agents.
Cultured patient-derived tumor fragments variably responded to urelumab plus nivolumab combinations
The potential showed by mouse-derived cultured tumor fragments to reveal the activity of a combination of immunotherapy agents would be of interest mainly if it allowed the interrogation of equivalent human tumor samples. We had access to a series of surgical specimens of endometrial, ovarian, and renal tumors following routine pathology diagnostic handling (Table S1).
Patient-derived tumor fragments were cultured similarly to the mouse counterparts, again omitting the extracellular matrix embedding of the fragments for reasons of simplicity. In the case of human samples, we tested the combination of nivolumab (anti-PD-1) with urelumab (agonist anti-CD137 mAb)Citation29 as schematized in .
Figure 5. Human tumor-derived cultured fragments in response to nivolumab (anti-PD-1) plus urelumab (anti-CD137 agonist).
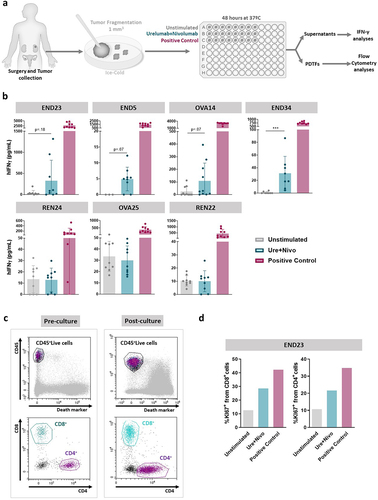
shows four cases in which the nivolumab plus urelumab combination led some of the fragments to release measurable levels of IFNγ into the supernatant and three cases in which no such activity could be detected, even if the PMA plus ionomycin positive control almost invariably induced IFNγ.
Of importance, the fragments retrieved following 48 h cultures contained viable CD8+ and CD4+ T cells as assessed by flow cytometry on single-cell suspensions (). Moreover, in one of the cases of endometrial carcinoma, more Ki67+ proliferating T cells were observed in the nivolumab plus urelumab stimulated cultures, as compared to the negative control ().
We next verified that T cells and IFNγ secretion activity were preserved following freezing and thawing of the fragmented samples (Figure S6a). Indeed, viable T lymphocytes were recovered from the fragments in which surface CD25 and intracellular Ki67 and granzyme B could be immunostained as shown in a representative case (Figure S6b) both in CD4+ and CD8+ lymphocytes. In a case of ovarian carcinoma, we observed that the anti-TGF-β (1D11 mAb) and the anti-TGF-β plus nivolumab plus urelumab triple combination readily increased the activation of T cells from the frozen/thawed fragments (Figure S6c). Moreover, the induction of IFNγ release into the supernatant as induced by the triple combination was preserved following freezing/thawing of the fragments prior to culture (Figure S6d).
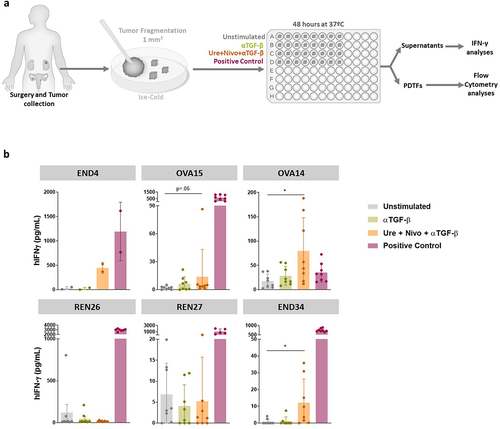
Triple combinations encompassing anti-CD137 plus anti-PD-1 plus anti-TGF-β tested on frozen/thawed and fresh cultured patient-derived tumor fragments
Once the freezing and thawing procedure was shown to be compatible with the intended experimentation, we tested six additional cases upon culture with nivolumab plus urelumab plus an anti-TGF-β mAb in a triple combination (), that our experiments in mice suggested to be synergistic.Citation28 We were able to substantiate some enhanced production of IFNγ into the supernatant in four of the 48 h cultures, while the other two gave no signal (). These results indicate that at least to some extent and in some cases combinations of experimental immunotherapy agents can be tested ex vivo for signs of immune cell activation in cancer tissues derived from diseases whose immunotherapy treatment outcomes are suboptimal at the present point of time.
Figure 6. Nivolumab plus urelumab plus anti-TGF-β stimulate IFNγ secretion in a fraction of tumor-derived fragments from cancer patients.
Discussion
This work set out to explore the possibility of ex-vivo testing immunotherapy combinations in small fragments derived from freshly excised tumor tissue. For this purpose, we focused on the combinations of anti-CD137 (4-1BB) agonists with PD-1 blocking agents. Such a combination is of interest following excellent results in preclinical modelsCitation19 and promising efficacy in the clinic.Citation15,Citation30
For proof of concept, we used mouse models in which the combination attains complete or partial efficacy to eradicate established tumors. Fragments from these murine engrafted tumors did show signs of increased production of IFNγ from tumor-infiltrating T lymphocytes in the treated mice. Importantly, we were able to retrieve cell suspensions containing viable T lymphocytes at the end of 72 h cultures on which flow cytometry assessments could be made.
The parameter that reproducibly could be measured in such murine tumor-derived fragments upon culture with immunostimulating antibodies was IFNγ, a cytokine that is increasingly recognized as one of the key mediators of anticancer immunityCitation31,Citation32 and one of the most responsive cytokines upon anti-PD-1 monotherapy exposure ex vivo.Citation7 Having said this, the system shows heterogeneity in the IFNγ response among mice and among fragments. Such heterogeneity on the ≈1 mm3 fragments probably reflects an uneven spatial distribution of immune cells within fragments.Citation7,Citation33 Tissue digestion might allow more homogeneous tumor-derived spheroidsCitation34 to be obtained at the expense of disrupting the tumor architecture that might be important for some heterologous immune-tumor cell interactions.
It is tempting to speculate that the most informative parameter perhaps is not the average of IFNγ released into the supernatant by each fragment but that achieved by top-scoring fragments of those tested. Further research should establish the optimal way to gain valuable translational information.
In light of the mouse experiments, we considered two interesting paths forward: (i) perhaps we could use the system to predict antitumor activity and (ii) perhaps we could identify other targets that, if concurrently or sequentially acted upon, will enhance the performance of the fragments in the cultures suggesting potential therapeutic synergies.
First, we set up bilateral tumors in the to-be-treated mice in which only one of the tumors was excised to analyze the ex-vivo response of its fragments to the immunostimulating antibodies and the other one was left in place for the in-vivo response evaluation to the subsequent treatment with the same antibodies. To our surprise, the correlation of IFNγ production from the fragments and tumor shrinkage in the MC38 tumor model was weak or non-existant. Many factors could account for this, since the cultures do not replicate many of the in-vivo tissue conditions such as the critical role of cross-priming of CD8+ T cells by dendritic cells conceivably taking place in lymph nodes.Citation35 Second, upon testing of an array of soluble mediators released into the supernatant, IL-4 was identified as being differentially produced by responding and non-responding cases. Given that IL-4 neutralization reportedly potentiates checkpoint inhibitors,Citation24 we tested the triple anti-PD-1 plus anti-CD137 plus anti-mouse IL-4 combination against established MC38 tumors, but only a marginal improvement was attained in terms of antitumor responses. We also tested anti-TGF-β, that in our hands and elsewhere in the literature is synergistic with checkpoint inhibitorsCitation25,Citation26,Citation28 and indeed could be detected in the tumor-fragment supernatants. In our settings, we were able to observe similar increased levels of IFNγ upon adding TGF-β neutralization to anti-PD-1 plus anti-CD137 treatment. The mouse models also permitted the validation of freezing and thawing procedures that were considered crucial to translate the strategy to human samples.
In the experimentation with human-derived fragments, we followed in the footsteps of D. Thommen’s group at the NKI.Citation7,Citation8 However, we did not include the fragments in collagen since mouse and human preliminary experiments did not show any meaningful differences in cytokine and flow cytometry readouts. We considered that we could reasonably simplify the culture technique without significantly influencing results provided the supernatant was carefully collected to prevent dragging the immune-rich cellular component that typically spills from tumor fragmentsCitation8,Citation36 and thereby preserve it for flow cytometry analysis.
Other authors use ex-vivo cultures of thin sections of tumor tissue prepared with a vibratome.Citation37 This constitutes an alternative but is technically more demanding. Another alternative but also technically more challenging approach are tumor-derived organoids, where pharmacodynamic changes induced by anti-PD-1 treatment have been described in mouse and patient-derived three-dimensional cultures.Citation4,Citation38,Citation39
In human samples, we observed remarkable levels of inter- and intra-patient heterogeneity, but importantly some levels of induction of IFNγ upon culture with the combination of nivolumab plus urelumab could be observed in a proportion of cases. Other authors have employed a similar method to test checkpoint inhibitors in combination with IL-2Citation10 or COX2 inhibitors.Citation9 Our results are suggestive of the fact that, in humans, anti-PD-1 plus anti-CD137 doublets and anti-PD-1 plus anti-CD137 plus anti-TGF-β1 triplets could be active in a proportion of low- to moderately immunogenic tumors. In the clinic, galunisertib, a small-molecule TGFβ-RI inhibitor, has been tested combined with chemotherapy and anti-PD-(L)1 agents, but results have been discouraging both in immunologically cold and responsive tumors.Citation40–42 Bintrafusp alfa, an innovative bifunctional protein composed of a monoclonal antibody against PD-L1 fused to the extracellular domain of TGF-βRII, showed preliminary efficacy in a phase 1 clinical trial,Citation43 but not in larger clinical trials.Citation44,Citation45 This advocates for potential combinations with CD137 agonists. Overall, data highlight that a better understanding of the TGF-β pathway in the TME is required to identify patients that might benefit from its blockade. Ex vivo tumor fragment cultures, with a preserved tumor architecture, could aid as biomarkers for the identification of responders in the future.
Regarding CD137 agonism, novel drug designs intended to overcome the dose-limiting liver toxicity of monoclonal antibodies by selectively targeting the TME are successfully completing early clinical trials.Citation12 The efficacy of CD137 agonism on TILs isolated from human cancer samples has already been described by the induction of effector molecules by CD8+ TILs.Citation46,Citation47 In addition, CD137-stimulated TILs during culture have been studied in a small pilot clinical trial.Citation48
One of the important limitations of ex vivo cultures is that their use is limited to a short time frame. In addition, it is obvious that tumor fragments are not exposed to the continuous arrival of new immune cells from peripheral blood as they are in vivo. This might be a limitation considering the accumulating evidence that suggests that ICB might act preventing exhaustion on newly arriving TILs rather than reverting terminal exhaustion in tumor-resident T cells.Citation49 Nevertheless, the concordance reported between ex vivo pharmacodynamic scores and clinical responses to anti-PD-1 therapy suggests that the TME contains the components required to initiate a short-term immune response induced by anti-PD-1 agents.Citation7
The fact that patient-derived tumor fragments can be frozen and thawed offers the opportunity to repeatedly test informative cases with different treatments and combinations. In this way, relatively simple procedures can be implemented to gather clinically relevant translational information from mouse models to human samples. The experimental system conveys useful information, with limitations, for the development of more active immunotherapy combinations encompassing doublets and beyond.
List of abbreviations
| ConA | = | concanavalin A. |
| COX2 | = | Cyclooxygenase-2. |
| CTLA-4 | = | cytotoxic T-Lymphocyte antigen 4. |
| DMSO | = | dimethylsulfoxide. |
| EDTA | = | ethylenediamine tetraacetic acid. |
| ELISA | = | enzyme-linked immunosorbent assay. |
| FACS | = | fluorescence-activated cell sorting. |
| FcR | = | Fc receptor. |
| IFNγ | = | interferon gamma. |
| IL-2 | = | interleukin 2. |
| IL-4 | = | interleukin 4. |
| MFI | = | mean fluorescence intensity. |
| NKI | = | National Cancer Institute (Netherlands). |
| PD-1 | = | programed death 1. |
| PHA | = | phytohemagglutinin. |
| PMA | = | phorbol 12-myristate 13-acetate. |
| TGF-β | = | transforming growth factor beta. |
| TILs | = | tumor-infiltrating lymphocytes. |
Author contributions
Project conception: I. E.-S., I. R., M. F. S and I. M.
Experiments: I.E.-S., I. R., C. H.-M., E. F.-P., A. A., S. S.-G., E. B. and A. T.
Human sample obtention: E. C., J. A. M., S. C, F. D.-C. and C. A.
Data interpretation: I. E.-S., I. R., C. H.-M., E. F.-P., A. A., S. S.-G., E. B., G. G., P. M.-G., E. C., J. A. M., S. C, F. D.-C., C. A., A. T, M. F.-S. and I. M.
Manuscript writing: I. E.-S., I. R. and I. M.
Manuscript review and editing: I. E.-S., I. R., C. H.-M., E. F.-P., A. A., S. S.-G., E. B., G. G., P. M.-G., E. C., J. A. M., S. C, F. D.-C., C. A., A. T, M. F.-S. and I. M.
Supplemental Material
Download Zip (1.1 MB)Supplemental Material
Download MS Word (28.1 KB)Acknowledgments
We are sincerely grateful to the patients who kindly consented to provide the human samples used in this project. We are thankful to Susana Chocarro Martin for her kind collaboration with patient identification.
Disclosure statement
I. E.-S., I. R., C. H.-M., E. F.-P., A. A., S. S.-G., E. B., G. G., P. M.-G., E. C., J. A. M., S. C, F. D.-C., C. A. and A. T. declare no conflicts of interest. M. F. S. reports grants from Bristol Myers Squibb and Roche during the conduct of the study, as well as grants and personal fees from Roche and Bristol Myers Squibb, and personal fees from Numab outside the submitted work. I. M. reports grants and personal fees from Genmab during the conduct of the study, as well as grants and personal fees from Bristol Myers Squibb, Roche, AstraZeneca, and Pharmamar and personal fees from F-Star, Numab, Pieris, Boehringer Ingelheim, Gossamer, Alligator, Hotspot, Biolinerx, Bioncotech, Dompe, Highlight Therapeutics, Bright Peaks and Boston Therapeutics outside the submitted work.
Data availability statement
Data are available upon reasonable request to corresponding authors.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2024.2373519
Additional information
Funding
References
- Perez-Gracia JL, Labiano S, Rodriguez-Ruiz ME, Sanmamed MF, Melero I. Orchestrating immune check-point blockade for cancer immunotherapy in combinations. Curr Opin Immunol. 2014;27(1):89–14. doi:10.1016/J.COI.2014.01.002.
- Melero I, Berman DM, Aznar MA, Korman AJ, Gracia JLP, Haanen J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer. 2015;15(8):457–472. doi:10.1038/nrc3973.
- Chen DS, Mellman I. Elements of cancer immunity and the cancer–immune set point. Nature. 2017;541(7637):321–330. doi:10.1038/nature21349.
- Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, Liu IH, Chiou SH, Salahudeen AA, Smith AR, et al. Organoid modeling of the tumor immune microenvironment. Cell. 2018;175(7):1972–1988.e16. doi:10.1016/J.CELL.2018.11.021.
- Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, Slagter M, van der Velden DL, Kaing S, Kelderman S, et al. Generation of tumor-reactive T Cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell. 2018;174(6):1586–1598.e12. doi:10.1016/J.CELL.2018.07.009.
- Chalabi M, Fanchi LF, Dijkstra KK, Van den Berg JG, Aalbers AG, Sikorska K, Lopez-Yurda M, Grootscholten C, Beets GL, Snaebjornsson P, et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med. 2020;26(4):566–576. doi:10.1038/s41591-020-0805-8.
- Voabil P, de Bruijn M, Roelofsen LM, Hendriks SH, Brokamp S, van den Braber M, Broeks A, Sanders J, Herzig P, Zippelius A, et al. An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nat Med. 2021;27(7):1250–1261. doi:10.1038/s41591-021-01398-3.
- Roelofsen LM, Voabil P, de Bruijn M, Herzig P, Zippelius A, Schumacher TN, Thommen DS. Protocol for ex vivo culture of patient-derived tumor fragments. Star Protoc. 2023;4(2):102282. doi:10.1016/J.XPRO.2023.102282.
- Pelly VS, Moeini A, Roelofsen LM, Bonavita E, Bell CR, Hutton C, Blanco-Gomez A, Banyard A, Bromley CP, Flanagan E, et al. Anti-inflammatory drugs remodel the tumor immune environment to enhance immune checkpoint blockade efficacy. Cancer Discov. 2021;11(10):2602–2619. doi:10.1158/2159-8290.CD-20-1815.
- Kaptein P, Jacoberger-Foissac C, Dimitriadis P, Voabil P, De Bruijn M, Brokamp S, Reijers I, Versluis J, Nallan G, Triscott H, et al. Addition of interleukin-2 overcomes resistance to neoadjuvant CTLA4 and PD1 blockade in ex vivo patient tumors. Sci Transl Med. 2022;14(642). doi:10.1126/SCITRANSLMED.ABJ9779.
- Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellström KE, Mittler RS, Chen L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med. 1997;3(6):682–685. doi:10.1038/nm0697-682.
- Melero I, Sanmamed MF, Glez-Vaz J, Luri-Rey C, Wang J, Chen L. CD137 (4-1BB)-based cancer immunotherapy on its 25th anniversary. Cancer Discov. 2023;13(3):552–569. doi:10.1158/2159-8290.CD-22-1029.
- Heumann T, Judkins C, Li K, Lim SJ, Hoare J, Parkinson R, Cao H, Zhang T, Gai J, Celiker B, et al. A platform trial of neoadjuvant and adjuvant antitumor vaccination alone or in combination with PD-1 antagonist and CD137 agonist antibodies in patients with resectable pancreatic adenocarcinoma. Nat Commun. 2023;14(1). doi:10.1038/S41467-023-39196-9.
- Tolcher AW, Sznol M, Hu-Lieskovan S, Papadopoulos KP, Patnaik A, Rasco DW, Di Gravio D, Huang B, Gambhire D, Chen Y, et al. Phase Ib Study of utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in combination with pembrolizumab (MK-3475) in patients with advanced solid tumors. Clin Cancer Res. 2017;23(18):5349–5357. doi:10.1158/1078-0432.CCR-17-1243.
- Muik A, Garralda E, Altintas I, Gieseke F, Geva R, Ben-Ami E, Maurice-Dror C, Calvo E, Lorusso PM, Alonso G, et al. Preclinical characterization and phase I trial results of a bispecific antibody targeting PD-L1 and 4-1BB (GEN1046) in patients with advanced refractory solid tumors. Cancer Discov. 2022;12(5):1248–1265. doi:10.1158/2159-8290.CD-21-1345.
- Sanmamed MF, Berraondo P, Rodriguez-Ruiz ME, Melero I. Charting roadmaps towards novel and safe synergistic immunotherapy combinations. Nat Cancer. 2022;3(6):665–680. doi:10.1038/s43018-022-00401-1.
- Morales-Kastresana A, Sanmamed MF, Rodriguez I, Palazon A, Martinez-Forero I, Labiano S, Hervas-Stubbs S, Sangro B, Ochoa C, Rouzaut A, et al. Combined immunostimulatory monoclonal antibodies extend survival in an aggressive transgenic hepatocellular carcinoma mouse model. Clin Cancer Res. 2013;19(22):6151–6162. doi:10.1158/1078-0432.CCR-13-1189.
- Ochoa MC, Sanchez-Gregorio S, de Andrea CE, Garasa S, Alvarez M, Olivera I, Glez-Vaz J, Luri-Rey C, Etxeberria I, Cirella A, et al. Synergistic effects of combined immunotherapy strategies in a model of multifocal hepatocellular carcinoma. Cell Rep Med. 2023;4(4):101009. doi:10.1016/j.xcrm.2023.101009.
- Azpilikueta A, Agorreta J, Labiano S, Pérez-Gracia JL, Sánchez-Paulete AR, Aznar MA, Ajona D, Gil-Bazo I, Larrayoz M, Teijeira A, et al. Successful immunotherapy against a transplantable mouse squamous lung carcinoma with Anti–PD-1 and Anti-CD137 monoclonal antibodies. J Thorac Oncol. 2016;11(4):524–536. doi:10.1016/J.JTHO.2016.01.013.
- Labiano S, Palazón A, Bolaños E, Azpilikueta A, Sánchez-Paulete AR, Morales-Kastresana A, Quetglas JI, Perez-Gracia JL, Gúrpide A, Rodriguez-Ruiz M, et al. Hypoxia-induced soluble CD137 in malignant cells blocks CD137L-costimulation as an immune escape mechanism. Oncoimmunology. 2015;5(1):e1062967. doi:10.1080/2162402X.2015.1062967.
- Palazón A, Martínez-Forero I, Teijeira A, Morales-Kastresana A, Alfaro C, Sanmamed MF, Perez-Gracia JL, Peñuelas I, Hervás-Stubbs S, Rouzaut A, et al. The HIF-1α hypoxia response in tumor-infi ltrating T lymphocytes induces functional CD137 (4-1BB) for immunotherapy. Cancer Discov. 2012;2(7):608–623. doi:10.1158/2159-8290.CD-11-0314.
- Glez-Vaz J, Azpilikueta A, Olivera I, Cirella A, Teijeira A, Ochoa MC, Alvarez M, Eguren-Santamaria I, Luri-Rey C, Rodriguez-Ruiz ME, et al. Soluble CD137 as a dynamic biomarker to monitor agonist CD137 immunotherapies. J Immunother Cancer. 2022;10(3):e003532. doi:10.1136/JITC-2021-003532.
- Martinez-Forero I, Azpilikueta A, Bolaños-Mateo E, Nistal-Villan E, Palazon A, Teijeira A, Perez-Chacon G, Morales-Kastresana A, Murillo O, Jure-Kunkel M, et al. T cell costimulation with anti-CD137 monoclonal antibodies is mediated by K63–polyubiquitin-dependent signals from endosomes. The J Immunol. 2013;190(12):6694–6706. doi:10.4049/JIMMUNOL.1203010.
- LaMarche NM, Hegde S, Park MD, Maier BB, Troncoso L, Le Berichel J, Hamon P, Belabed M, Mattiuz R, Hennequin C, et al. An IL-4 signalling axis in bone marrow drives pro-tumorigenic myelopoiesis. Nature. 2024;625(7993):166–174. doi:10.1038/S41586-023-06797-9.
- Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Cañellas A, Hernando-Momblona X, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–543. doi:10.1038/NATURE25492.
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE, Koeppen H, Astarita JL, Cubas R, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–548. doi:10.1038/NATURE25501.
- Massagué J, Sheppard D. TGF-β signaling in health and disease. Cell. 2023;186(19):4007–4037. doi:10.1016/J.CELL.2023.07.036.
- Rodríguez-Ruiz ME, Rodríguez I, Mayorga L, Labiano T, Barbes B, Etxeberria I, Ponz-Sarvise M, Azpilikueta A, Bolaños E, Sanmamed MF, et al. TGFβ blockade enhances radiotherapy abscopal efficacy effects in combination with anti-PD1 and Anti-CD137 immunostimulatory monoclonal antibodies. Mol Cancer Ther. 2019;18(3):621–631. doi:10.1158/1535-7163.MCT-18-0558.
- Segal NH, Logan TF, Hodi FS, McDermott D, Melero I, Hamid O, Schmidt H, Robert C, Chiarion-Sileni V, Ascierto PA, et al. Results from an integrated safety analysis of urelumab, an agonist anti-CD137 monoclonal antibody. Clin Cancer Res. 2017;23(8):1929–1936. doi:10.1158/1078-0432.CCR-16-1272.
- Melero I, Tanos T, Bustamante M, Sanmamed MF, Calvo E, Moreno I, Moreno V, Hernandez T, Garcia MM, Rodriguez-Vida A, et al. A first-in-human study of the fibroblast activation protein-targeted, 4-1BB agonist RO7122290 in patients with advanced solid tumors. Sci Transl Med. 2023;15(695). doi:10.1126/SCITRANSLMED.ABP9229.
- Thibaut R, Bost P, Milo I, Cazaux M, Lemaître F, Garcia Z, Amit I, Breart B, Cornuot C, Schwikowski B, et al. Bystander IFN-γ activity promotes widespread and sustained cytokine signaling altering the tumor microenvironment. Nat Cancer. 2020;1(3):302–314. doi:10.1038/S43018-020-0038-2.
- Larson RC, Kann MC, Bailey SR, Haradhvala NJ, Llopis PM, Bouffard AA, Scarfó I, Leick MB, Grauwet K, Berger TR, et al. CAR T cell killing requires the IFNγR pathway in solid but not liquid tumours. Nature. 2022;604(7906):563–570. doi:10.1038/S41586-022-04585-5.
- de Witte CJ, Espejo Valle-Inclan J, Hami N, Lõhmussaar K, Kopper O, Vreuls CPH, Jonges GN, van Diest P, Nguyen L, Clevers H, et al. Patient-derived ovarian cancer organoids mimic clinical response and exhibit heterogeneous inter- and intrapatient drug responses. Cell Rep. 2020;31(11):107762. doi:10.1016/J.CELREP.2020.107762.
- Jiang S, Zhao H, Zhang W, Wang J, Liu Y, Cao Y, Zheng H, Hu Z, Wang S, Zhu Y, et al. An automated organoid platform with inter-organoid homogeneity and inter-patient heterogeneity. Cell Rep Med. 2020;1(9):100161. doi:10.1016/J.XCRM.2020.100161.
- Sánchez-Paulete AR, Teijeira A, Cueto FJ, Garasa S, Pérez-Gracia JL, Sánchez-Arráez A, Sancho D, Melero I. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann Oncol. 2017;28(suppl_12):xii74. doi:10.1093/ANNONC/MDX727.
- Kamer I, Bab-Dinitz E, Zadok O, Ofek E, Gottfried T, Daniel-Meshulam I, Hout-Siloni G, Ben Nun A, Barshack I, Onn A, et al. Immunotherapy response modeling by ex-vivo organ culture for lung cancer. Cancer Immunol, Immunother. 2021;70(8):2223–2234. doi:10.1007/s00262-020-02828-w.
- Sivakumar R, Chan M, Shin JS, Nishida-Aoki N, Kenerson HL, Elemento O, Beltran H, Yeung R, Gujral TS. Organotypic tumor slice cultures provide a versatile platform for immuno-oncology and drug discovery. Oncoimmunology. 2019;8(12):e1670019. doi:10.1080/2162402X.2019.1670019.
- Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, Chhabra S, Huang W, Liu H, Aref AR, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 2018;8(2):216–233. doi:10.1158/2159-8290.CD-17-0915.
- Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, Bowden M, Deng J, Liu H, Miao D, et al. Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discov. 2018;8(2):196–215. doi:10.1158/2159-8290.CD-17-0833.
- Brandes AA, Carpentier AF, Kesari S, Sepulveda-Sanchez JM, Wheeler HR, Chinot O, Cher L, Steinbach JP, Capper D, Specenier P, et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol. 2016;18(8):1146–1156. doi:10.1093/NEUONC/NOW009.
- Melisi D, Oh DY, Hollebecque A, Calvo E, Varghese A, Borazanci E, Macarulla T, Merz V, Zecchetto C, Zhao Y, et al. Safety and activity of the TGFβ receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J Immunother Cancer. 2021;9(3):e002068. doi:10.1136/JITC-2020-002068.
- Nadal E, Saleh M, Aix SP, Ochoa-de-Olza M, Patel SP, Antonia S, Zhao Y, Gueorguieva I, Man M, Estrem ST, et al. A phase Ib/II study of galunisertib in combination with nivolumab in solid tumors and non-small cell lung cancer. BMC Cancer. 2023;23(1):1–13. doi:10.1186/s12885-023-11153-1.
- Strauss J, Heery CR, Schlom J, Madan RA, Cao L, Kang Z, Lamping E, Marte JL, Donahue RN, Grenga I, et al. Phase I Trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGFβ, in advanced solid tumors. Clin Cancer Res. 2018;24(6):1287–1295. doi:10.1158/1078-0432.CCR-17-2653.
- Cho BC, Lee JS, Wu YL, Cicin I, Dols MC, Ahn MJ, Cuppens K, Veillon R, Nadal E, Dias JM, et al. Bintrafusp Alfa versus pembrolizumab in patients with treatment-naive, programmed death-ligand 1-high advanced NSCLC: a randomized, open-label, phase 3 trial. J Thorac Oncol. 2023;18(12):1731–1742. doi:10.1016/J.JTHO.2023.08.018.
- Vokes EE, Mornex F, Sezer A, Cheng Y, Fang J, Baz DV, Cil T, Adjei AA, Ahn MJ, Barlesi F, et al. Bintrafusp Alfa with CCRT followed by Bintrafusp Alfa versus Placebo with CCRT followed by Durvalumab in Patients with Unresectable Stage III NSCLC: a phase 2 randomized study. J Thorac Oncol. 2024;19(2):285–296. doi:10.1016/J.JTHO.2023.09.1452.
- Leem G, Park J, Jeon M, Kim ES, Kim SW, Lee YJ, Choi SJ, Choi B, Park S, Ju YS, et al. 4-1BB co-stimulation further enhances anti-PD-1-mediated reinvigoration of exhausted CD39+ CD8 T cells from primary and metastatic sites of epithelial ovarian cancers. J Immunother Cancer. 2020;8(2):e001650. doi:10.1136/JITC-2020-001650.
- Shen Y, Li XL, Li YX, Shan ZG, Zhao YL, Cheng P, Zhao Z, Zhang JY, Chen W, Zhuang Y, et al. Distribution, phenotype, functional and clinical relevance of CD8+CD103+ tissue-resident memory T cells in human gastric cancer. Cancer Immunol, Immunother. 2022;71(7):1645–1654. doi:10.1007/s00262-021-03105-0.
- Amaria R, Knisely A, Vining D, Kopetz S, Overman MJ, Javle M, Antonoff MB, Tzeng CWD, Wolff RA, Pant S, et al. Efficacy and safety of autologous tumor-infiltrating lymphocytes in recurrent or refractory ovarian cancer, colorectal cancer, and pancreatic ductal adenocarcinoma. J Immunother Cancer. 2024;12(2):e006822. doi:10.1136/JITC-2023-006822.
- Mellman I, Chen DS, Powles T, Turley SJ. The cancer-immunity cycle: indication, genotype, and immunotype. Immunity. 2023;56(10):2188–2205. doi:10.1016/J.IMMUNI.2023.09.011.