
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The level of tumor and circulating CXCR1/2-expressing neutrophils and CXCR1/2 ligands correlate with poor patient outcomes, inversely correlate with tumoral lymphocyte content, and predict immune checkpoint inhibitor (ICI) treatment failure. Accordingly, CXCR2-selective and CXCR1/2 dual inhibitors exhibit activity both as single agents and in combination with ICI treatment in mouse tumor models. Based on such reports, clinical trials combining CXCR1/2 axis antagonists with ICI treatment for cancer patients are underway. It has been assumed that CXCR1/2 blockade impacts tumors by blocking neutrophil chemotaxis and reducing neutrophil content in tumors. Here, we show that while CXCR2 antagonism does slow tumor growth, it does not preclude neutrophil recruitment into tumor. Instead, CXCR1/2 inhibition alters neutrophil function by blocking the polarization of transcriptional programs toward immune suppressive phenotypes and rendering neutrophils incapable of suppressing lymphocyte proliferation. This is associated with decreased release of reactive oxygen species and Arginase-1 into the extracellular milieu. Remarkably, these therapeutics do not impact the ability of neutrophils to phagocytose and kill ingested bacteria. Taken together, these results mechanistically explain why CXCR1/2 inhibition has been active in cancer but without infectious complications.
Introduction
Neutrophils, or polymorphonuclear (PMN) cells, are the most abundant white blood cell type in peripheral blood. They are the innate immune system’s first line of defense and a primary component of the inflammatory response. The recruitment of neutrophils to sites of infection and inflammation is essential for the neutralization and removal of bacterial, fungal, and viral pathogens.Citation1 Resolution of inflammation requires the diminution of neutrophils and other immune cells to basal levels, which involves the elimination of their chemotactic gradients.Citation2 However, in chronic inflammatory processes such as cancer, neoplastic cells continue to secrete a variety of chemokines and cytokines that mediate perpetual waves of neutrophil recruitment.Citation3 Hence, the analogy that cancers represent wounds that do not heal.Citation4 Neutrophil recruitment to sites of inflammation and tumorigenesis is facilitated by redundant chemotactic signals that include leukotriene B4 (LTB4), complement 5a (C5a), and several CXC ligands. CXCR1 (only expressed in humans) and CXCR2 (expressed in mice and humans) are G protein coupled receptors that constitute the primary mechanism for neutrophil chemotaxis and recruitment.Citation5,Citation6 These potent receptor–ligand axis is shown to be elevated in several inflammatory diseases, including chronic obstructive pulmonary disease (COPD), asthma, rheumatoid arthritis, psoriasis, inflammatory bowel disease, and cancer.Citation7–10
Although there exist reports of both pro-host and pro-tumor functions of tumor-associated neutrophils (TAN) in humans,Citation11 recent reports, including one demonstrating that neutrophil infiltration predicts immune checkpoint inhibitor (ICI) treatment failure in non-small cell lung cancer (NSCLC),Citation12–14 have led to the performance of clinical investigations in mice employing neutrophil antagonists, frequently in combination with ICI.Citation15–25 Based on these results, human clinical trials have been initiated. Specifically, agents targeting CXCR2 selectively (AZD5069), CXCR1/CXCR2 (SX-682), and the CXCR1/CXCR2 ligand CXCL8/IL-8 (BMS-986253) are being studied in patients with lung cancer (NCT04123379 and NCT05570825), pancreatic cancer (NCT02583477 and NCT04477343), melanoma (NCT03161431), and other solid cancers (NCT04599140), in combination with anti-PD(L)1 antibody therapies. The general assumption with this therapeutic strategy is that CXCR1/CXCR2 axis blockade will limit neutrophil chemotaxis and recruitment such that the neutrophil content with the TME will be reduced. To that end, the typical readout for on target activity of these drugs is reduced TAN content in an on-treatment biopsy or the development of neutropenia, the latter of which has been used to push therapeutic doses to the highest possible levels. Given that there several alternate mechanisms that drive neutrophil migration (e.g. complement products, eicosanoid lipid mediators, collagen fragments, etc.), we suspected that targeting the CXCR2 axis alone would be unlikely to yield a near-complete exclusion of neutrophils within tumors in vivo. Therefore, we undertook a series of experiments designed to assess the impact of CXCR1/CXCR2 antagonism on neutrophil content and function in lung cancer models in mice. Here, we show that two different CXCR2 antagonists blunt tumor growth without reducing TAN content in mice. Instead, the immune suppressive effects of TAN are inhibited while sparing the ability of neutrophils to phagocytose and kill bacteria within the neutrophil phagolysosome.
Materials and methods
Mice and in vivo treatments
Lewis lung carcinoma cells that were obtained from ATCC and were cultured in complete DMEM with 10% heat-inactivated FBS and penicillin/streptomycin (1 × 105) were injected into the parenchyma of the left lung lobe through the rib cage, as described previously in the gender and age matched C57BL/6 mice.Citation26 B6.129S4-Ptentm1Hwu/J (Ptenfl/fl) and Stk11tm1.1Sjm/J (Lkb1fl/fl) mice on pure C57BL/6 backgrounds were acquired from The Jackson Laboratory. Ptenfl/fl; Lkb1fl/fl (PL) mice were generated by simple crossbreeding and have been previously reported. PL mice received an intratracheal dose of 1 × 107 PFU of adenoviral Cre recombinase (AdCre; University of Iowa Viral Vector Core, Iowa City, Iowa, USA) to initiate tumorigenesis between 8 and 10 weeks of age, as previously described.Citation27 Tumor-bearing lungs were confirmed and their volumes (W × L × D) were calculated using μCT 10 d post implantation for LLC and 30–35 weeks post infection for PL mice. CXCR2 antagonist (AZD; 100 mg/kg) and CXCR1/CXCR2 antagonist (SX-682; 200 mg/kg) were given twice daily o.p. Anti-Ly6G (1A8; BP0075–1) and anti-rat Kappa immunoglobulin light chain antibodies (MAR18.5; BE0122) administered as described in Boivin et al.Citation28 C57BL.6-Arg1tm1Pmu/J (Arg1fl/fl) mice were all obtained from The Jackson Laboratory. Ly6G-Cre mice were provided by Matthias Gunzer (University Duisburg-Essen, Essen, Germany).Citation29 The Ly6G-Cre; Arg1fl/fl mice were generated by simple crossbreeding and have been previously reported. NE−/− mice were generated by targeted gene disruption as previously detailed.Citation30
Tissue processing and flow cytometry
Single-cell suspensions were prepared from tumor-bearing lungs by mechanically dissociating the tissue with scissors and digesting in collagenase IV (Worthington), 80 U/mL DNase (Worthington), and Pierce protease inhibitor cocktail (ThermoSci; A32963) at 37c for 30 min. The resulting cells were incubated with mouse TruStain FcX (Biolegend) for 15 min on ice and stained for 30 min at room temperature with either the flow antibody panel #1 or #2. Flow antibody panel #1: CD4 (GK1.5; BUV563; BD), CD8 (53–6.7; FITC; BD), CD11b (M1/70; APC-R700; BD), CD11c (N418; BV421; Biolegend), CD64 (X54–5/7.1; PE-Cy7; BD), CD45 (30-F11; BUV661; BD), CD3 (17A2; BV750; Biolegend), MHCII (M5/114; BV711; BD), Ly6C (RA3–6B2; BV570; eBioscience), and with Live/Dead Fixable Blue (Thermosci) to assess viability. Flow antibody panel #2: CD4 (GK1.5; BUV563; BD), PDL1 (MIH5;BUV615; BD), CD45 (30-F11;BUV661; BD), Ly6G (1A8; BUV737; BD), CD39 (24DMS1; BUV805; BD), CD11c (N418;BV421; Biolegend), Ly6C (HK1.4; BV570; Biolegend), PD1 (J43;BV605;BD), XCR1 (ZET; BV650; BD), MHCII (M5/114.15.2; BV711; Biolegend), CD8a (53–6.7; BB515; BD), CXCR3 (CXCR3–173; NFB610; Invitrogen), TCRβ (H57–597; BB700; BD), CD172a (P84; PerCPe710; BD), CD73 (eBioTY/11.8; PE; eBioscience), CD64 (X54–5/7.1; PE-Cy7; Biolegend), CD11b (M1/70; APC-R700; BD), CD38 (90; APC-Fire810; Biolegend), and Live/Dead Fixable Blue (Thermosci) to assess viability. Samples were analyzed using the BD FACSymphony II cell analyzer or Sony ID7000 analyzer for panel #1 and #2, respectively. Compensation and gating analysis for the panel #1 were done with FlowJo Software (Tree Star).
The flow gating strategy is provided in . Of note, for this study, neutrophils were defined as CD11b+Ly6G+ cells. For the unique case of measuring neutrophil content in mice that had received Ly6G depleting antibody, we utilized an alternate method because the Ly6G flow antibodies are the same clone as the neutrophil depleting antibody. For this specific case, we defined neutrophils as CD11b+Ly6Cint cells.
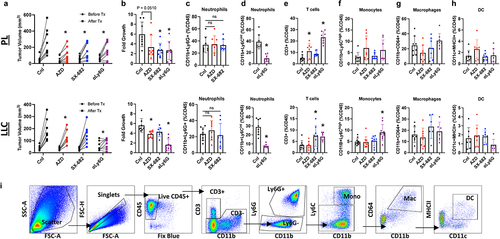
Figure 1. CXCR1/2 antagonists blunt tumor growth without reducing tumor-associated neutrophil content in mice. The results of autochthonous PL lung squamous model and orthotopic LLC model are shown on the top and bottom row, respectively. (A-B) lung tumor burden is expressed in tumor volume (WxLxD) and fold changes measured with uCT scans before and after 2 weeks of the indicated treatments. AZD and SX-682 was given P.O. 100 mg/kg or 200 mg/kg twice daily, respectively. To deplete neutrophils, mice were given 200 mg/kg of anti-Ly6G (1A8) daily and anti-rat IgGk every other day I.P. for 2 weeks. (C-D) neutrophils (CD11b+Ly6G+ or CD11b+Ly6Cint), (E) lymphocytes (CD3+), (F) monocytes (CD11b+Ly6Chi), (G) macrophages (CD11b+CD64+), and (H) dendric cells (DC; CD64−CD11c+MHCII+) were characterized using flow cytometry and expressed as the percentages of total live CD45. Each dot represents a single tumor-bearing mouse from 3 separate experiments, n = 8, mean s.d. ordinary one-way ANOVA with Tukey’s post hoc test *p < 0.05. (I) The representative flow cytometry gating strategy is shown.
Immunohistochemistry (IHC)
IHC staining and MSI was performed using PerkinElmer’s Opal fIHC reagents and the Aperio VERSA200 Imaging System (Leica Biosystems) as described in Kargl et al.Citation14 Anti-mouse Ly6G (1A8; Biolegend 127,602) was used at 1:2000 and Opal 570 was used at 1:80 dilution.
Boyden cell migration assay
Tumor lysates (1 mg/mL) or mouse recombinant mouse CXCL2 (50 ng/mL; R&D; 452-M2) were prepared in HBSS and used in neuroprobe cell migration chamber with 3-µm filter. Mice were treated with vehicle control or either CXCR2 antagonist for more than 2 d prior to inducing sterile peritonitis by injecting 1 mL of 4% thioglycolate intraperitoneally. Peritoneal neutrophils were collected from peritoneal exudates 4 h after the induction of sterile peritonitis. Hemocytometer counts and modified Wright stained cytospins were utilized to confirm >85% neutrophil purity. The cells were used in the migration chamber in HBSS at 2 × 106 cells/mL. After 3 h, the migrated cells were counted by flow cytometry.
ELISA
Mouse CXCL1/KC Quantikine ELISA kit (R&D; MKC00B), CXCL2/MIP-2 Quantikine ELISA kit (R&D; MM200), Complement C5a mouse ELISA kit (Invitrogen; EMHC), and Leukotriene B4 Express ELISA kit (Cayman; 10009292) were used according to manufacturer’s protocol on tumor lysates that were prepared in PBS and Pierce protease inhibitor tablet (ThermoSci; A32963). BCA assay was used to ensure equal loading.
Single cell RNA-sequencing
Tumor-associated neutrophils were isolated using fluorescence-assisted cell sorting (FACS) of the live Ly6G population from single-cell suspensions of tumor-bearing lungs that have been stained with anti-Ly6G (1A8; BUV737, BD) and LIVE/DEAD Fixable Blue Dead Cell Stain Kit for UV excitation for viability on Sony MA900 cell sorter into FBS containing PBS. Cells were loaded onto the 10X Genomics Chromium Controller using the Chromium Next GEM Single Cell 3’v3.1 kit to partition the cells into nanoliter-scale single-cell Gel beads-in-emulsion (GEMs). Then, the GEMs were placed in the thermocycler for the reverse transcription of the polyadenylated mRNA. cDNA was amplified to generate Gene Expression libraries. The libraries were sequenced in the illumina NextSeq2000 sequencer using a P3–100 flow cell, following the 10X sequencing recommendations: Read1 28 cycles, Read2 90 cycles, index1 10 cycles and index2 10 cycles. The final pool was loaded at 1200 pM and 1% PhiX illumina control was spiked in. Single-cell RNA sequencing data for tumor-associated neutrophils were analyzed using the CELLxGENE.
T cell proliferation assay
TAN were isolated with mouse anti-Ly6G MicroBeads UltraPure (130-120-337: Miltenyi Biotec) kit according to the manufacture’s recommendation. Splenic T cells were isolated with mouse EasySeo T cell isolation kit (Stemcell; 19851) from naïve C57BL/6 mice and were stimulated in T cell medium (RPMI 1640, penicillin/streptomycin, 200 mM l-glutamine, 50 mM β-mercaptoethanol, 1 M HEPES) with L-arginine (75 μM) and recombinant human 50 U/mL of IL-2 (Peprotech). Dynabeads mouse T-activator CD3/CD28 beads were used according to manufacturer’s protocol. The cells were incubated at indicated ratio for overnight before the T cell activating beads were removed. The T cells and TANs were returned to the tissue incubator for 24 h before the CellTrace violet signal was measuring using flow cytometer.
Apoptosis detection
Cells were incubated in annexin-binding buffer with the annexin V conjugates at 1:200 dilution and Live/Dead Fixable Blue (Thermosci; L23105) at 1:800 dilution for 15 min at the room temperature. The cells were washed and analyzed by flow cytometry.
Arginase I activity assay
Activities of Arginase I of 2 × 105 cells were measured using ARG1 activity assay kit (Abcam; ab180877) according to the manufacturer’s protocol. For conditioned media, the cells were incubated directly in the plates. Whole cell lysates were made with cell lysis buffer (Cell signaling tech; #9803) and loaded into the activity assay. The manufacturer’s protocol was followed.
NE activity assay
NE was determined by measurement of p-nitroanilide resulting from hydrolysis of the neutrophil elastase-specific peptide, N-methoxysuccinyl-Ala-Ala-Pro-Val-p-nitroanilide. Human NE isolated from human sputum leukocyte elastase (SPC; SE562) was used to create the standard, ranging from 1 μM to 1 × 10−5 μM. 2 × 105 cells per well was used for either conditioned media or whole cell lysates. Conditioned media was made by incubating 2 × 105 cells in 100 μL of PBS for 1 h. The cell lysates were made with 1× cell lysis buffer with 2 × 105 cells. The manufacturer’s protocol was followed.
Arginase I ELISA
The total arginase I content was measured using mouse Arginase I ELISA kit (ab269541). Conditioned media was made by incubating 2 × 105 cells in 100ul of PBS for 1 h. The cell lysates were made with 1× cell extract buffer provided by the kit with 2 × 105 cells. The manufacture’s protocol was followed.
2‘,7’-Dichlorodihydrofluorescein diacetate (H2DCFDA) and 4-amino-5-methylamino-2’7’-difluororescein (DAF-FM) assay
Cells were loaded with 5 μM H2DCFDA (Invitrogen; D399) or 5 μM DAF-FM (Invitrogen; D23841) for 15 min at 37°C in PBS, and the manufacturer’s protocol was followed.
Immunofluorescence and confocal imaging
Cells (2 × 106 cells/mL) were activated with 250 nM of phorbol myristate acetate (PMA) for 3 h on a glass-bottom 96-well plate. Following the induction of NETosis, the reaction was terminated with 4% (w/v) paraformaldehyde (PFA) overnight, permeabilized with 1% Triton X-100 for 25 min and blocked with 2.5% (w/v) BSA in PBS overnight. Citrulline Histone 3 (H3Cit) was probed for using mouse anti-H3Cit antibody (AbboMax: 630–180; clone:nan) at 1:150 dilution. DAPI (100 μM, Thermo Fisher Scientific) at 1:333 dilution was used for visualizing DNA. A Dragonfly 200 high-speed confocal imaging platform (Andor Technology) was mounted on a Leica DMi8 (Leica Microsystems) with a 100X/1.4 NA objective lens under the control of Fusion version 2.3.0.36 (Oxford Instruments). Images were captured using 488-nm and/or 561-nm lasers with an Andor iXon Life 888 EMCCD camera (Andor Technology) with the 2X mag changer. The analysis of the acquired images was done using Imaris software; The spots algorithm was chosen to identify H3Cit based on GFP signal. Appropriate DAPI signal threshold was set and applied to all images to categorize the spots into intra or extra nuclear H3Cit.
Phagocytosis assay
Cells were incubated with pHrodo Green E. coli BioParticles or pHrodo Red S. aureus BioParticles (P35366 and A10010) for 15 min at 37°C. The phagocytic capacities were compared using a flow cytometry to measure the percentage and mean fluorescence intensities of GFP or PE positive cells, respectively.
Bactericidal assay
The indicated bacteria (1 × 107 cells/mL) were incubated with the cells (2 × 106 cells/mL) for 15 min at 37°C. The wells were gently washed with gentamycin (100 μg/mL) and separated into two time points: 0 min and 30 min. The 0 min cells were lysed after the gentamycin wash, whereas the 30 min cells were incubated in PBS for 30 min before lysed. For both timepoints, the cells were lysed with 0.1% Triton X and appropriate dilutions were made before plating onto LB plates. After overnight, CFU was calculated, 30 min timepoint data was subtracted from the 0 min timepoint and divided from the 0 min timepoint data to measure the percentage of bactericidal capacities.
Experimental pneumonia
Mice of each treatment and control were treated for 4 d prior to infection. On the infection day, 50 μl of S. aureus and E. coli at 2 × 107 and 4 × 107 pfu/mL, respectively, into the tracheas of mice. Bronchial Alveolar Lavage (BAL) samples were collected by injecting and recovering 1 mL of PBS from the lungs of the mice after 18 h. One hundred microliters of each BAL fluid was plated to calculate the bacterial clearance.
Statistical methods
Data are expressed as the mean ± standard deviation (s.d.) unless otherwise stated. Statistical analyses were performed on Prism 10 software. Multiple comparisons were assessed using 1-way ANOVA with Tukey’s post hoc test. A 2-way ANOVA was used specifically to assess differences between tumor volume pairs. p Values of less than 0.05 were considered statistically significant.
Study approval
All animal experiments used age- and sex-matched mice and were conducted at the Fred Hutchison Cancer Center using protocols approved by the Institutional Animal Care and Use Committee.
Results
CXCR1/2 antagonists blunt tumor growth without reducing tumor-associated neutrophil content in mice
We assessed the impact on tumor burden induced by anti-Ly6G antibody mediated neutrophil depletion as compared to that accomplished by administration of the CXCR2-selective inhibitor, AZ12376429–026 (murine version of AZD5069 which will be referred to as AZD in this study) and the dual CXCR1/2 inhibitor, SX-682. To accomplish this, lung tumors in both the Ptenfl/flLkb1fl/fl (PL) autochthonous mouse model of lung squamous cell carcinoma (LUSQ) and the Lewis Lung Carcinoma (LLC) orthotopic model of lung adenocarcinoma (LUAD) were imaged by μCT, and tumors of comparable sizes were randomly assigned into the treatment or control groups. AZD or SX-682 as single agents showed modest, but significant, inhibition on the terminal tumor burden and the rate of tumor growth expressed in fold growth over 2 weeks of treatment in both mouse models, respectively (). The importance of neutrophils in tumor progression was further highlighted by the fact the tumor growth was most stunted ( ; purple) by depleting neutrophils using a combination of anti-Ly6G (1A8) and anti-rat IgGκ as described in Boivin et al.Citation28 (). To our surprise, neither AZD nor SX-682 induced a significant reduction in TAN content when expressed either by the percentage of CD45 () or the total cells per milligram of tissue (Supplemental Figure S1A), or by immunohistochemistry (Supplemental Figure S1G). Despite the lack of neutrophil cellular exclusion from the lung tumors, we observed a significant increase in percentages of CD3+ cells in tumor-bearing lungs that had been treated with AZD and SX-682 ( and Supplemental Figure S1B-C), though this was the most pronounced in both models with Ly6G antibody treatment (). The increased CD3+ cellular content was not accompanied by any significant changes in monocyte, macrophage, or dendritic cell (DC) content with the two antagonists (), except we did observe a significant increase in monocytes in the LLC model with neutrophil depletion, likely reflecting a reciprocal change (). To extend these findings, we repeated this experiment with a different flow panel that include CD4, CD8, and T cell activation markers such as CD39, CD38, CD73, PD1, and CXCR3. We were unable to identify significant changes in the percentages of CD39+CD73+, CD38+CD73+, PD1+, and CXCR3+ in CD4+ or CD8+ cells nor in the percentages of CD39+CD73+, CD38+CD73+, and PDL1+ in Ly6G+ and CD64+ positive cells (Supplemental Figure S1H and I). Taken together, we have concluded that CXCR2 antagonism inhibited tumor growth rates in two different lung cancer models without significantly reducing TAN content.
CXCR1/2 antagonism does not confer a loss of neutrophil chemotaxis
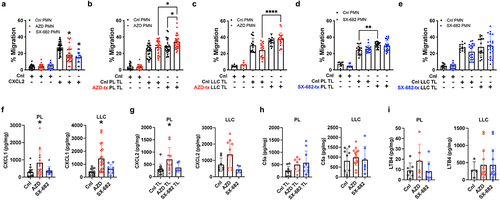
First, we assessed the effect of the CXCR1/2 antagonism on neutrophil migration toward recombinant CXCL2. In , Boyden cell migration assay showed significant, but incomplete inhibition of neutrophil migration toward the CXCL2 gradient (50 ng/mL) with either antagonist. Our objective here was not to titer the two antagonists in mice or the concentration of CXCL2 gradient to achieve near-complete inhibition of neutrophil migration, but to demonstrate that at a therapeutic dose (100 mg/kg for AZD and 200 mg/kg for SX-682), these two antagonists can indeed inhibit neutrophil migration toward CXCR2-dependent ligands – at least to an extent. However, when using tumor lysates (TL) from PL or LLC tumor-bearing lungs, which presumably contain the complex mixture of neutrophil chemotactic molecules encountered in vivo, the CXCR1/2 antagonists failed to significantly impede neutrophil chemotaxis (, comparing either the third and fourth bar, or fifth and sixth bar). Surprisingly, the capacity to attract neutrophils was further enhanced when either LLC or PL tumors were treated with AZD (comparing the fourth and sixth bars in ). In contrast, SX-682 neither increased nor decreased the chemotactic capacities of PL and LLC tumors (). The enhanced capacity of AZD-treated LLC and PL tumors to attract neutrophils is partly explained with upregulated levels of CXCL1 and CXCL2, the prototypical ligands for CXCR2 (). However, the increases were only observed in tumors that were treated with AZD, but not with SX-682, suggesting possible differences in compensation mechanisms elicited by the two antagonists. Furthermore, the levels of the chemotactic agents C5a and LTB4 were not impacted by CXCR1/2 axis inhibition (), but the synergistic nature of the multiple permutations of different chemokines remains unknown. Taken together, we concluded that AZD and SX-682 inhibit CXCR2-CXCL2-mediated chemotaxis to an extent; however, multiple compensatory mechanisms likely exist in vivo.
Figure 2. CXCR1/2 antagonism does not confer a loss of neutrophil chemotactic capacities of tumors. Following a 4-d treatment period (black = control, red = AZD, and blue = SX-682), thioglycolate-induced neutrophils (>85% pure) were isolated from the peritoneum exudates and used in the modified Boyden chamber chemotaxis assay. The chemotactic capacities of the indicated neutrophils were assessed toward the gradients by (A) CXCL2 (50 ng/mL), (B and C) AZD-treated PL and LLC tumor lysates, and (D-E) SX-682-treated PL and LLC tumor lysates. Each closed circle represents an experimental replicate from three separate biological replicates with the indicated tumor lysates (1 mg/mL) prepared from at least three different mice. The mean percentages of migrated neutrophil over total seeded cells s.d. Are shown. (F-I) CXCL1, CXCL2, C5a, and LTB4 contents in whole tumor lysates of the indicated tumor model and treatment were measured by ELISA and expressed as pg/mg of total protein loaded. Each open circle represents a lysate of the tumor bearing lung of mouse that were treated with vehicle control, AZD, or SX-682 for 2 weeks. The graphs show the mean
s.d., and ordinary one-way ANOVA with Tukey’s post hoc test (*p < 0.05) was used for the statistical analysis.
CXCR1/2 antagonism perturbs neutrophil polarization into pro-tumor sub-phenotypes
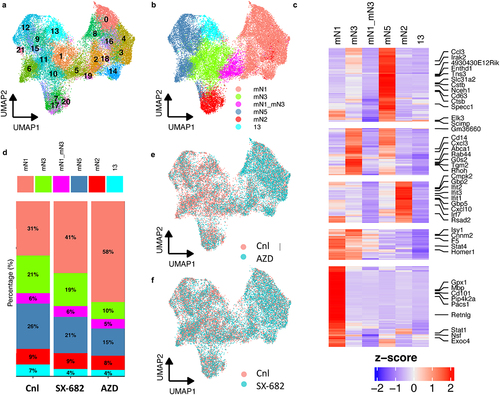
Since the CXCR1/2 antagonists reduced tumor burden rate without a significant exclusion of neutrophils, we suspected that these agents were impacting neutrophil function, and not recruitment. To test this in an unbiased way, we used scRNA-seq to analyze the effect of CXCR1/2 antagonism on the live Ly6G+ cells in the lungs of LLC tumor-bearing mice. Unsupervised analysis of scRNA-seq revealed 21 unique clusters based on their gene expression (). Previously, Zilionis and colleagues identified 6 neutrophil clusters in mice and 5 in human samples.Citation31 They classified the cluster that expressed Mmp8, Mmp9, S100A8 and S100A9, and Adam8 as mN1, or canonical neutrophils. They further subdivided the neutrophils into groups mN3 through mN5 based on the expression of genes such as Ccl3, Csf1, Cstb, Ctsb, and Irak2. These observations fit the narrative that TAN progressively polarize from the mN1 canonical state to more immunosuppressive and pro-tumor phenotypes (mN3–mN5). The authors also identified a subtype that is characterized by expression of Ifng-inducible genes (mN2) that is disparate from the other groups and believed to represent a pro-host TAN subgroup. To visualize the polarization of neutrophil clusters in our dataset, we annotated our clusters within the context of published gene sets and were able to identify the mN1, mN2, mN3, and mN5 populations. However, we were not able to identify cluster mN4 or mN6 – instead, we saw a hybrid cluster of mN1 and mN3 (mN1_mN3) and a unique cluster 13 (). Upon closer examination of cluster composition, we found that the mN1 cluster was proportionally larger in the antagonist groups compared to control, suggesting that CXCR1/2 inhibition renders canonical neutrophils (mN1) incapable of polarizing into more tumor promoting phenotypes (). In addition, the proportion of mN2 cluster remained constant among all the groups, reaffirming that there might be a constant TAN population that are sensitive to the Ifng signal. The shift from mN1 to mN3 and mN5 is best visualized in the UMAP plots comparing AZD- and SX-682-treated TANs with control TANs (). Each green dot representing a single TAN from either antagonist dominates the right-hand side of the UMAP, whereas the red dots dominate the left-hand side of the UMAP. Thus, it appears that CXCR1/2 antagonism perturbs TAN transcriptional profiles in favor of a canonical state as opposed to immune suppressive phenotypes.
Figure 3. CXCR1/2 antagonism perturbs neutrophil polarization into pro-tumor sub-phenotypes. Live, Ly6G+ cells from the LLC lung tumor-bearing mice were used. Each dot represents a single TAN. (A) UMAP plots 21 clusters of neutrophils identified by the single-cell RNA-sequencing. (B) UMAP plot of the annotated clusters based on gene signatures previously reported. (C) Heatmap showing genes upregulated (red) and downregulated (blue) in TAN clusters. Negative and positive Z-scores respectively associate with increased or decrease expression. (D) The population compositions in percentages of whole are calculated for the annotated clusters for each treatment group. (E and F) UMAP plots comparing the polarities of control versus AZD and SX-682 TANs, respectively.
CXCR1/2 antagonists eliminate the suppressive effects of TAN on T cell proliferation in vitro
Prior studies of CXCR2-deficient myeloid cells have shown that CXCR2 signaling may play an important role in the ability of neutrophils to produce potentially immune suppressive substances (e.g., ROS).Citation18,Citation32 This suggests that CXCR2 function is not limited to chemotaxis and may bear functional significance. Therefore, we tested the hypothesis that the gene expression changes identified in the sc-RNA-seq studies above would impart changes in neutrophil function with respect to lymphocyte suppression. To accomplish this, we used TANs isolated from mice that were treated with a CXCR1/2 antagonist for at least 4 d. Naïve splenic T cells were stained with CellTrace Violet and seeded with anti-CD3/CD28 beads and the indicated TANs at 1:1:3 (bead:T cell:TAN) ratio. The sequential loss of CellTrace Violet signal among the proliferating T cells indicated that over 80% and 60% of the CD4+ and CD8+ T cells were dividing without exposure to TANs, respectively. However, when incubated with control TAN, they were significantly inhibited from proliferating (). To our surprise, the neutrophils that had been treated with either AZD or SX-682 had completely lost their ability to suppress CD8+ T cells, whereas their loss of suppressive effects on CD4+ T cells were significant but not complete (). The same observation was made when we used higher T cells to TAN ratios – both AZD and SX-682 TANs showed significant defect in inhibiting T cell proliferation compared to the control TANs (Supplemental Figure S2A). In addition, we ruled out the possibility of CXCR1/2 antagonism inducing early apoptosis or death of the neutrophils. To address this, we incubated the neutrophils in full-serum T cell media that we used for the above experiment and measured the apoptotic and dead cells every 24 h. Supplemental Figure S2B shows that there are no significant differences among the percentage live, apoptotic cells, or dead cells in CXCR1/2 antagonized neutrophils. Overall, this evidence strongly supports the hypothesis that the CXCR1/2 signaling plays a crucial role in mediating the suppressive effects of neutrophils on T cell proliferation.
Figure 4. CXCR1/2 antagonists limit the suppressive function of PMN on T cell proliferation in vitro. TAN were purified using the anti-Ly6G positive selection from the LLC tumors treated with the indicated treatments for 4 d. The TANs were incubated with T cells that were stained with CellTrace violet (1 μM) and anti-CD3/CD28 beads at the 1:1:3 (T cells: beads: TANs) ratio. (A) The proliferating T cells were measured by the loss of CellTrace violet signal and expressed as percentage of total CD4 or CD8 T cells. The representative histograms of CellTrace violet signal of T cells incubated with the indicated TANs are shown. (B and C) Arg1 activity and NE activity, respectively, were measured in the conditioned media (CM; circles) and whole cell lysates (WCL; squares) of neutrophils with the indicated treatment. Neutrophils isolated from Arg1 or NE knockout mice were used as negative control for the respective activity assays. (D and E) generation of reactive oxygen intermediates or nitric oxide was measured in the neutrophils that were loaded with cell-membrane permeable 2’7’-dichlorodihydroflourescin diacetate (H2DCFDA) or 4-amino-5-methylamino-2’7’-difluororescein (DAF-FM), respectively. (F) Neutrophils with the indicated treatment were allowed to attach to the glass bottom 96-well plate and activated with 250 nM of PMA for 3 h. Following the induction of NETosis, citrulline histone 3 (H3Cit; green) was probed, and DAPI (blue) was used for visualizing nucleus. The graph shows the ratios of the extranuclear over intranuclear H3Cit spots in neutrophils with indicated treatment. Representative images acquired by confocal microscope are shown in either the XY or Z coordinates with or without bright field (gray). The graphs show the mean s.d., and ordinary one-way ANOVA with Tukey’s post hoc test (*p < 0.05) was used for the statistical analysis. Each dot represents a replicate from three separate biological experiments. Graphs show the mean
s.d., and ordinary one-way ANOVA with Tukey’s post hoc test (*p < 0.05) was used for the statistical analysis.
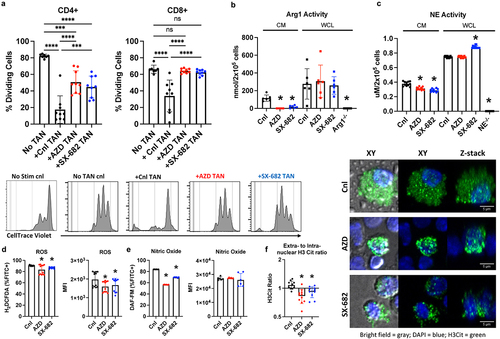
To gain deeper mechanistic insight, we measured arginase-1 (Arg1) activity and neutrophil elastase (NE) activity in the conditioned media (CM) and whole cell lysates (WCLs) of neutrophils.Citation30 shows that CXCR1/2 blockade significantly reduced the ability of the neutrophils to convert L-arginine into urea or to hydrolyze N-methoxysuccinyl-Ala-Ala-Pro-Val p-nitroanilide (MSAPN) – a peptide used to measure neutrophil elastase (NE)-specific activity – in the conditioned media, but not in the whole cell lysates (WCL). The comparable total Arg1 cellular content was confirmed in the WCLs with Arg1 ELISA (Supplemental Figure S3A). These observations suggest that the CXCR1/2 antagonists-treated neutrophils still possess ample intracellular content of Arginase I and NE, but are unable to release these effector molecules into the extracellular space. In addition, the levels of the reactive oxygen species (ROS) and nitric oxide (NO) were measured using the cell permeable fluorescent probe, 2‘,7’-dichlorodihydrofluorescein diacetate (H2DCFDA) and 4-amino-5-methylamino-2’7’-difluororescein (DAF-FM), respectively. The result suggested modest reductions in reactive oxygen intermediates and nitric oxide in the CXCR1/2 inhibitor-treated neutrophils (). Arg1 and ROS are two well-known mediators of lymphocyte suppression, and the significant reductions in these two key components are possible mechanisms by which CXCR1/2 antagonism can limit the pro-tumor functions of neutrophils.
Lastly, we examined the effects of CXCR1/2 antagonism on the formation of neutrophil-extracellular traps (NETs) in neutrophils. NETosis is a unique mechanism by which neutrophils execute their antimicrobial and pro-tumorigenic functions.Citation33,Citation34 It has been shown to inhibit staphylococcal skin infection extravasation into the bloodstream and to aid in cancer metastasis.Citation35 To assess NETosis, we used immunofluorescence (IF) to measure extranuclear citrulline Histone 3 (H3Cit) by contrasting with nuclear DAPI staining. Compared to the control, we found significantly lower ratios of extranuclear or intranuclear H3Cit, suggesting that the CXCR1/2 antagonists may hinder NETosis (). It has been previously shown that ROS generation is a critical step in NETosis and dependent of JAK2-dependent ERK phosphorylation.Citation36,Citation37 However, we were able to detect any significant changes in the phosphorylation status of ERK1/2 or AKT (Supplemental Figure S3B). Although we do not yet understand the signaling defect with the CXCR1/2 antagonism, the data, overall, suggest that CXCR1/2 ligands modify essential roles in mediating neutrophil behaviors within the TME.
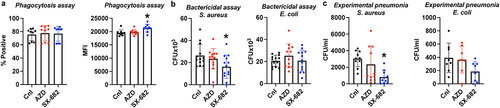
CXCR1/2 antagonism does not impact phagocytosis and intracellular bacterial killing by neutrophils
The crucial role of neutrophils in the fight against bacterial infection cannot be overstated. Therefore, the concern over the use of CXCR1/2 antagonists in cancer patients that they might be prone to infections is not unwarranted, especially given our observation that NETosis and NE release may be defective in the neutrophils whose CXCR1/2 signaling pathway has been targeted. To test the effects of CXCR2 antagonism on the phagocytic capacity of neutrophils, we incubated neutrophils with pHrodo BioParticles. These beads are non-fluorescent outside the cell at neutral pH but fluoresce when phagocytosed into endosome. shows no defect in the neutrophils’ ability to phagocytose the beads in numbers or degree measured by the percentage positive or MFI, respectively, with either the CXCR2 antagonist. The pitfall of this assay is that increased phagocytosis does not necessarily mean bacterial clearance. Therefore, we incubated live, log-phase E. coli and S. aureus with neutrophils after which the neutrophils were washed with gentamycin to eliminate any adherent or associated extracellular bacteria. The cells were incubated in PBS for 30 min, then lysed, and plated onto LB plates. The results indicated that neither antagonist reduced the bactericidal activities of neutrophils against S. aureus or E. coli (). These results should not be surprising since NE and ROS are well-known to exert their antimicrobial effects within the neutrophil phagolysosome and do not require extracellular release (which may be inhibited by CXCR1/2 antagonists) for bacterial killing. To provide a better clinical context, we instilled S. aureus and E. coli (in separate experiments) into the lungs of mice that had been treated with one of the antagonists (or vehicle) starting 4 d prior to infection. After 18 h, bronchial alveolar lavage (BAL) specimens were plated to compare the bacterial loads. The resulting colony forming units (CFUs) of the BAL from mice treated with AZD were not significantly different to that of the control, suggesting that the antagonist did not reduce the host immune system from clearing bacterial infection. Surprisingly, the pneumonia model indicated that SX-682 significantly enhances the host immune system to clear infection (). The mechanisms underlying the enhanced bactericidal capacity by SX-682 treatment are not readily apparent. It is important to note that we did not titrate up the doses of either antagonists to the point of neutropenia in our mouse models. It is certainly possible that at higher dose, the bactericidal capacities of neutrophils are impacted. Further studies delineating the role of CXCR2 antagonism in the process of neutrophil-mediated bacterial killing is currently underway in our laboratory. However, we have concluded that these therapeutic doses of CXCR2 antagonists do not impart an increased risk of infection, consistent with the initial results from formal clinical investigations.Citation38
Figure 5. CXCR1/2 antagonism does not impact phagocytosis and intracellular bacterial killing by neutrophils. (A) pHrodo BioParticles conjugates for phagocytosis were incubated with neutrophils that were treated with the indicated CXCR1/2 antagonist. The phagocytotic capacities of the neutrophils are expressed by percentage positive cells and MFI after 15 min of incubated at 37C and measured by flow cytometry. (B) The capacity of neutrophils to kill live S. aureus or E. coli, respectively, were examined as previously described by Belaaouaj et al. (1998).Citation39 The neutrophils were allowed 30 min after unassociated bacteria were washed off with gentamycin. The cells were then lysed and plated – the resulting colony forming unit (CFU) represents bacteria that have been phagocytosed, but not yet killed. (C) The bacterial burdens from the experimental pneumonia study are expressed in CFU/mL in the bronchial alveolar lavage (BAL) collected from the mice infected with S. aureus or E. coli, respectively. The infected mice were allowed to clear the indicated bacteria for overnight before BAL were collected. Each dot represents a mouse or experimental replicate from three separate biological experiments. The graphs show the mean s.d., and ordinary one-way ANOVA with Tukey’s post hoc test (*p < 0.05) was used for the statistical analysis.
Discussion
Neutrophils are often abundant within the TME, inversely correlate with lymphocyte content, and associate with ICI treatment failures.Citation12,Citation14,Citation40 These observations have been supported by several studies performed using mouse models of cancer demonstrating pro-tumor roles for TAN and that their depletion or inhibition reduces tumor growth.Citation15,Citation41 Accordingly, clinical trials have emerged employing neutrophil antagonizing therapies, most frequently in combination with immune checkpoint blockade. Most of these trials have employed CXCR2, CXCR1/2, or IL-8 antagonists, all of which were designed to block the dominant chemotactic axis for neutrophil recruitment thereby limiting their recruitment into the TME. Having studied many of these therapeutics in our laboratory, we repeatedly observed that they did, in fact, reduce tumor burden but surprisingly did not reduce TAN content. Therefore, we undertook a series of experiments designed to identify their impact. Here, we show for the first time that CXCR1/2 blockade inhibits neutrophil function but not recruitment within the TME.
In retrospect, it should not be surprising that inhibition of the CXCR1/2 axis does not abolish neutrophil recruitment to the TME. Likely because of their importance in combating invading microorganisms, neutrophil chemotactic factors are highly redundant and include several factors in addition to CXCR1/2 ligands.Citation2 The in vitro chemotactic assays performed here show that CXCR1/2 blockade does, in fact, inhibit neutrophil chemotaxis to CXCR1/2 ligands, such as recombinant CXCL2. However, the complicated mixture of neutrophil chemoattractants that exist within the TME (harbored within the tumor lysates used here experimentally) was fully capable of inducing neutrophil chemotaxis despite CXCR1/2 blockade. Although we did not find a significant upregulation of C5a or LTB4 in our lysates, there are numerous other mediators of neutrophil chemotaxis that could be involved, such as CCL4, a ligand for CCR5.Citation42,Citation43 Identification of the operative ligand(s) using unbiased approaches is an active area of investigation in our laboratory.
The results presented here support two interrelated roles for CXCR1/2 blockade in altering neutrophil function. First, it is clear from the sc-RNA-seq data that neutrophil transcriptional profiles have been altered and point toward an inhibition of polarization toward more tumor-promoting phenotypes. This implies that the activity of certain pathways or production of certain proteins have been impacted. As an example, intracellular ROS activity is reduced within anti-CXCR1/2-treated neutrophils. Second, the ability of neutrophils to release effector molecules has been impacted in a way that does not implicate protein or effector molecule content. For example, Arg-1 intracellular protein content (by ELISA) and function (by Arg1 activity assay) were not affected by CXCR1/2 blockade. However, Arg1 activity was reduced by CXCR1/2 blockade in the extracellular space, where its presence would be required to suppress lymphocyte proliferation. Similarly, NE intracellular granule content is not affected by CXCR1/2 blockade though NE activity in the extracellular space was reduced. These results suggest that neutrophil function has been impacted by polarization of transcriptional profile and content of certain proteins and by the inability to release some effector molecules into the extracellular space in response to CXCR1/2 signaling.
CXCR1/2 antagonism does not appear to affect phagocytosis nor the ability of neutrophils to kill ingested bacteria in vitro and in vivo. In fact, phagocytosis was modestly increased with SX-682, which is of unclear etiology and significance at this time. The reduction in ROS and NO production alluded to above could theoretically reduce the ability of neutrophil to kill bacteria within the phagolysosome. Neutrophils possess numerous other anti-bacterial entities (NE, defensins, lipocalin, etc.) that may compensate for the reduction of ROS production.Citation44 Alternatively, ROS supply may still reach satisfactory intra-phagolysosome levels to accomplish bacterial killing.
One notable limitation of our study is the reliance on the study of mouse neutrophils and the absence of experiments utilizing human neutrophils. This is the result of the technical limitations associated with using human neutrophils. Although most of the assays utilized here can be satisfactorily performed using human neutrophils, the short ex vivo lifespan of neutrophils precludes ex vivo administration of study drugs for enough time to impact their function. In the future, we plan to repeat these studies using neutrophils isolated from patients receiving SX-682 as part of a clinical trial, such that these neutrophils would be devoid of CXCR1/2 signaling. We should also discuss the fact that our group previously reported a decrease in TAN content induced by SX-682 in the PL model.Citation14 In that study, we only used Ly6G IHC to measure TAN content and from a single section. Therefore, we repeated the IHC studies here (Supplemental ) and reviewed slides from the prior study. We observed that the neutrophil content is anatomically sporadic and simply counting a limited number of high-power fields would not be an accurate measure of total tumor TAN content. The IHC data presented here did not identify differences in TAN content induced by CXCR1/2 axis inhibition, consistent with the flow cytometry data.
The results reported here provide much needed insight as to the mechanisms by which CXCR1/2 inhibitors alter neutrophil function and recruitment in the setting of cancer therapy. Many of the ongoing clinical trials mentioned in the opening are relying on on-treatment biopsy neutrophil content to indicate on-target activity of the CXCR1/2 axis antagonist. Based on the results presented here, this outcome is likely not achievable. Instead, correlative studies examining the sequelae of blocking TAN function and its inherent lymphocyte suppression, such as increased infiltration of CD8+ T cells and restoration of the IFNG signature, may prove superior biomarkers of drug activity. Lastly, since abrogation of neutrophil immune suppressive functions may be observed at doses lower than those that induce neutropenia, a more refined therapeutic window may be possible by further exploration of clinical dose–response relationships.
Author contributions
JWK designed and performed experiments, crafted the figures, and wrote the manuscript. HQN, AC, GMH, SM, NNK, and XZ performed experiments and interpreted data. AMH conceived of the study, provided funding, interpreted data, and edited the manuscript.
Supplemental Figures.pdf
Download PDF (1.5 MB)Acknowledgments
The authors would like to acknowledge assistance from Fred Hutch Shared Resources (supported by NIH grant 5P30CA015704-49), especially the directors, staff scientists, and technicians at the Flow Cytometry, Experimental Histopathology, Cellular Imaging, Comparative Medicine, and Genomics and Bioinformatics core. Special thanks to Dean Maeda and John Zebala at Syntrix Pharmaceuticals for supplying SX-682 and to AstraZeneca for supplying AZ12376429-026. This work was supported by grants from the NIH to AMH (R01CA221295, R01CA282465, and P50CA228944). Author contributions: JK designed and performed the experiments and wrote the paper. AMH conceptualized and oversaw experiments and wrote the paper. All other authors participated in data acquisition and analysis. None of the authors have any conflicts of interest.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2024.2384674
Additional information
Funding
References
- Mocsai A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J Exp Med. 2013;210(7):1283–11. doi:10.1084/jem.20122220.
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–175. doi:10.1038/nri3399.
- Ji H, Houghton AM, Mariani TJ, Perera S, Kim CB, Padera R, Tonon G, McNamara K, Marconcini LA, Hezel A. et al. K-ras activation generates an inflammatory response in lung tumors. Oncogene. 2006;25(14):2105–2112. doi:10.1038/sj.onc.1209237.
- Dvorak HF. Tumors: wounds that do not heal-redux. Cancer Immunol Res. 2015;3(1):1–11. doi:10.1158/2326-6066.CIR-14-0209.
- Richardson RM, Pridgen BC, Haribabu B, Ali H, Snyderman R. Differential cross-regulation of the human chemokine receptors CXCR1 and CXCR2. Evidence for time-dependent signal generation. J Biol Chem. 1998;273(37):23830–23836. doi:10.1074/jbc.273.37.23830.
- Baggiolini M, Dewald B, Moser B. Human chemokines: an update. Annu Rev Immunol. 1997;15(1):675–705. doi:10.1146/annurev.immunol.15.1.675.
- Strydom N, Rankin SM. Regulation of circulating neutrophil numbers under homeostasis and in disease. J Innate Immun. 2013;5(4):304–314. doi:10.1159/000350282.
- Zhang W, Wang H, Sun M, Deng X, Wu X, Ma Y, Li M, Shuoa SM, You Q, Miao L. et al. CXCL5/CXCR2 axis in tumor microenvironment as potential diagnostic biomarker and therapeutic target. Cancer Commun (Lond). 2020;40(2–3):69–80. doi:10.1002/cac2.12010.
- Bullock K, Richmond A. Suppressing MDSC recruitment to the tumor microenvironment by antagonizing CXCR2 to enhance the efficacy of immunotherapy. Cancers (Basel). 2021;13(24):6293. doi:10.3390/cancers13246293.
- Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, Buczkowski KA, Liu Y, Awad MM, Denning WL. et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016;76(5):999–1008. doi:10.1158/0008-5472.CAN-15-1439.
- Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer. 2016;16(7):431–446. doi:10.1038/nrc.2016.52.
- Kargl J, Busch SE, Yang GH, Kim KH, Hanke ML, Metz HE, Hubbard JJ, Lee SM, Madtes DK, McIntosh MW. et al. Neutrophils dominate the immune cell composition in non-small cell lung cancer. Nat Commun. 2017;8(1):14381. doi:10.1038/ncomms14381.
- Ocana A, Nieto-Jimenez C, Pandiella A, Templeton AJ. Neutrophils in cancer: prognostic role and therapeutic strategies. Mol Cancer. 2017;16(1):137. doi:10.1186/s12943-017-0707-7.
- Kargl J, Zhu X, Zhang H, Yang GHY, Friesen TJ, Shipley M, Maeda DY, Zebala JA, McKay-Fleisch J, Meredith G. et al. Neutrophil content predicts lymphocyte depletion and anti-PD1 treatment failure in NSCLC. JCI Insight. 2019;4(24). doi:10.1172/jci.insight.130850.
- Steele CW, Karim SA, Leach JD, Bailey P, Upstill-Goddard R, Rishi L, Foth M, Bryson S, McDaid K, Wilson Z. et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell. 2016;29(6):832–845. doi:10.1016/j.ccell.2016.04.014.
- Sun L, Clavijo PE, Robbins Y, Patel P, Friedman J, Greene S, Das R, Silvin C, Van Waes C, Horn LA. et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight. 2019;4(7). doi:10.1172/jci.insight.126853.
- Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, Li J, Wang G, Lan Z, Li J. et al. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell. 2019;35(4):559–72 e7. doi:10.1016/j.ccell.2019.02.008.
- Jamieson T, Clarke M, Steele CW, Samuel MS, Neumann J, Jung A, Huels D, Olson MF, Das S, Nibbs RJB. et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J Clin Invest. 2012;122(9):3127–3144. doi:10.1172/JCI61067.
- Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, Kaplan RN, Mackall CL. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. 2014;6(237):237ra67. doi:10.1126/scitranslmed.3007974.
- Dominguez C, Kk M, David JM, Palena C. Neutralization of IL-8 decreases tumor PMN-MDSCs and reduces mesenchymalization of claudin-low triple-negative breast cancer. JCI Insight. 2017;2(21). doi:10.1172/jci.insight.94296.
- Raccosta L, Fontana R, Maggioni D, Lanterna C, Villablanca EJ, Paniccia A, Musumeci A, Chiricozzi E, Trincavelli ML, Daniele S. et al. The oxysterol–CXCR2 axis plays a key role in the recruitment of tumor-promoting neutrophils. J Exp Med. 2013;210(9):1711–1728. doi:10.1084/jem.20130440.
- Greene S, Robbins Y, Mydlarz WK, Huynh AP, Schmitt NC, Friedman J, Horn LA, Palena C, Schlom J, Maeda DY. et al. Inhibition of MDSC trafficking with SX-682, a CXCR1/2 inhibitor, enhances NK-Cell immunotherapy in head and neck cancer models. Clin Cancer Res. 2020;26(6):1420–1431. doi:10.1158/1078-0432.CCR-19-2625.
- Gungabeesoon J, Gort-Freitas NA, Kiss M, Bolli E, Messemaker M, Siwicki M, Hicham M, Bill R, Koch P, Cianciaruso C. et al. A neutrophil response linked to tumor control in immunotherapy. Cell. 2023;186(7):1448–64 e20. doi:10.1016/j.cell.2023.02.032.
- Pfirschke C, Engblom C, Gungabeesoon J, Lin Y, Rickelt S, Zilionis R, Messemaker M, Siwicki M, Gerhard GM, Kohl A. et al. Tumor-promoting ly-6G(+) SiglecF(high) cells are mature and long-lived neutrophils. Cell Rep. 2020;32(12):108164. doi:10.1016/j.celrep.2020.108164.
- Mollaoglu G, Jones A, Wait SJ, Mukhopadhyay A, Jeong S, Arya R, Camolotto SA, Mosbruger TL, Stubben CJ, Conley CJ. et al. The lineage-defining transcription factors SOX2 and NKX2-1 determine lung cancer cell fate and shape the tumor immune microenvironment. Immunity. 2018;49(4):764–79 e9. doi:10.1016/j.immuni.2018.09.020.
- Nemenoff R. Activation of PPARγ in myeloid cells promotes lung cancer progression and metastasis. Oncoimmunology. 2012;1(3):403–404. doi:10.4161/onci.19309.
- Busch SE, Hanke ML, Kargl J, Metz HE, MacPherson D, Houghton AM. Lung cancer subtypes generate unique immune responses. The J Immunol. 2016;197(11):4493–4503. doi:10.4049/jimmunol.1600576.
- Boivin G, Faget J, Ancey PB, Gkasti A, Mussard J, Engblom C, Pfirschke C, Contat C, Pascual J, Vazquez J. et al. Durable and controlled depletion of neutrophils in mice. Nat Commun. 2020;11(1):2762. doi:10.1038/s41467-020-16596-9.
- Hasenberg A, Hasenberg M, Mann L, Neumann F, Borkenstein L, Stecher M, Kraus A, Engel DR, Klingberg A, Seddigh P. et al. Catchup: a mouse model for imaging-based tracking and modulation of neutrophil granulocytes. Nat Methods. 2015;12(5):445–452. doi:10.1038/nmeth.3322.
- Zhang H, Zhu X, Friesen TJ, Kwak JW, Pisarenko T, Mekvanich S, Velasco MA, Randolph TW, Kargl J, Houghton AM. et al. Annexin A2/TLR2/MYD88 pathway induces arginase 1 expression in tumor-associated neutrophils. J Clin Invest. 2022;132(22). doi:10.1172/JCI153643.
- Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, Krishnan I, Maroni G, Meyerovitz CV, Kerwin CM. et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity. 2019;50(5):1317–34 e10. doi:10.1016/j.immuni.2019.03.009.
- Delobel P, Ginter B, Rubio E, Balabanian K, Lazennec G. CXCR2 intrinsically drives the maturation and function of neutrophils in mice. Front Immunol. 2022;13:1005551. doi:10.3389/fimmu.2022.1005551.
- Adrover JM, Sac M, He XY, Quail DF, Egeblad M. NETworking with cancer: the bidirectional interplay between cancer and neutrophil extracellular traps. Cancer Cell. 2023;41(3):505–526. doi:10.1016/j.ccell.2023.02.001.
- Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, Schott AF, Kinugasa-Katayama Y, Lee Y, Won NH. et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med. 2016;8(361):361ra138. doi:10.1126/scitranslmed.aag1711.
- Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, Huang D, Li J, Li H, Chen F. et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. 2020;583(7814):133–138. doi:10.1038/s41586-020-2394-6.
- Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–241. doi:10.1083/jcb.200606027.
- Hurtado-Nedelec M, Csillag-Grange MJ, Boussetta T, Belambri SA, Fay M, Cassinat B. Increased reactive oxygen species production and p47phox phosphorylation in neutrophils from myeloproliferative disorders patients with JAK2 (V617F) mutation. Haematologica. 2013;98(10):1517–1524. doi:10.3324/haematol.2012.082560.
- Rennard SI, Dale DC, Donohue JF, Kanniess F, Magnussen H, Sutherland ER, Watz H, Lu S, Stryszak P, Rosenberg E. et al. CXCR2 antagonist MK-7123. A phase 2 proof-of-concept trial for chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;191(9):1001–1011. doi:10.1164/rccm.201405-0992OC.
- Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley TJ, Abraham SN, Shapiro SD Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat Med. 1998;4:615–618.
- Lizotte PH, Ivanova EV, Awad MM, Jones RE, Keogh L, Liu H, Dries R, Almonte C, Herter-Sprie GS, Santos A. et al. Multiparametric profiling of non–small-cell lung cancers reveals distinct immunophenotypes. JCI Insight. 2016;1(14):e89014. doi:10.1172/jci.insight.89014.
- Pekarek LA, Starr BA, Toledano AY, Schreiber H. Inhibition of tumor growth by elimination of granulocytes. J Exp Med. 1995;181(1):435–440. doi:10.1084/jem.181.1.435.
- Satpathy SR, Jala VR, Bodduluri SR, Krishnan E, Hegde B, Hoyle GW, Fraig M, Luster AD, Haribabu B. Crystalline silica-induced leukotriene B4-dependent inflammation promotes lung tumour growth. Nat Commun. 2015;6(1):7064. doi:10.1038/ncomms8064.
- Kwak JW, Laskowski J, Li HY, McSharry MV, Sippel TR, Bullock BL, Johnson AM, Poczobutt JM, Neuwelt AJ, Malkoski SP. et al. Complement activation via a C3a receptor pathway alters CD4(+) T lymphocytes and mediates lung cancer progression. Cancer Res. 2018;78(1):143–156. doi:10.1158/0008-5472.CAN-17-0240.
- Levy O. Antimicrobial proteins and peptides: anti-infective molecules of mammalian leukocytes. J Leukoc Biol. 2004;76(5):909–925. doi:10.1189/jlb.0604320.