
ABSTRACT
The CRISPR/Cas9 technology is a welcome breakthrough for genome editing, owing to its precision, efficiency, versatility and ease of adoption. We recently reported the first application of CRISPR/Cas9 for biallelic mutations in stably transformed Populus, extending the species range of this powerful technology to woody perennials. An underappreciated obstacle in genome editing of outcrossing species is the frequent occurrence of sequence polymorphisms that can render CRISPR/Cas9 unproductive. We discuss experimental evidence as well as genome-wide computational analysis to demonstrate the sensitivity of CRISPR/Cas9 to allelic heterozygosity, and highlight tools and strategies that can help deal with such sequence polymorphisms. With its specificity, CRISPR/Cas9 offers a less equivocal means than previous approaches for discerning functional redundancy of paralogous genes that are prevalent in plant genomes. Continuing improvements of the CRISPR/Cas9 system for multiplex genome engineering should facilitate these efforts. The paradigm shift brought about by CRISPR/Cas9 promises to accelerate not only basic research but also applied crop improvement progress.
ABBREVIATIONS
| 4CL | = | 4-coumarate:CoA ligase |
| Cas9 | = | CRISPR-associated protein 9 |
| CRISPR | = | clustered regularly interspaced short palindromic repeats |
| CT | = | condensed tannin |
| gRNA | = | guide RNA |
| M | = | million |
| PAM | = | protospacer adjacent motif |
| Pol III | = | RNA polymerase III |
| RNAi | = | RNA interference |
| RNase | = | ribonuclease |
| SNP | = | single nucleotide polymorphism |
| S/G lignin | = | syringyl-to-guaiacyl lignin ratio |
| tRNA | = | transfer RNA |
Since the first report of its programmable gene editing potential, the CRISPR/Cas9 technology is revolutionizing all facets of biology from medicine to agriculture (Jinek et al., Citation2012). With its efficiency and simplicity, CRISPR/Cas9 has quickly displaced its predecessors (e.g., Zinc finger and transcription activator-like effector nucleases) as the method of choice for genome editing (Carroll, Citation2014). For agricultural applications that have traditionally depended on gene-silencing for RNA modification, CRISPR/Cas9 is a game changer. The previous methods, such as antisense or RNA interference (RNAi), leave much to be desired as the degree, specificity and stability of gene silencing are not always predictable. This necessitates screening and characterization of a large number of transgenic lines for the desired trait. By contrast, CRISPR/Cas9 is guided to disrupt the reading frame—thereby protein function—of the target gene by DNA editing. DNA recognition and cleavage occurs on both strands (Jinek et al., Citation2012), and both monoallelic and biallelic mutations are reported (Bortesi and Fischer, Citation2015). With biallelic editing, null mutations are readily obtained in primary transformants, though with varying efficiencies (Brooks et al., Citation2014; Zhang et al., Citation2014; Zhou et al., Citation2014, Citation2015), Interestingly, the most efficient and consistent CRISPR/Cas9 editing was reported in hybrid Populus tremula x alba clone 717-1B4 (717), with biallelic mutations detected in all independent transgenic lines examined (Zhou et al., Citation2015). For woody perennials such as forest trees, fruit/nut trees and woody ornamentals with highly heterologous genomes and long generation times, CRISPR/Cas9 affords a facile means to accelerate genetic improvement.
HIGH-EFFICIENCY GENOME EDITING OF POPULUS
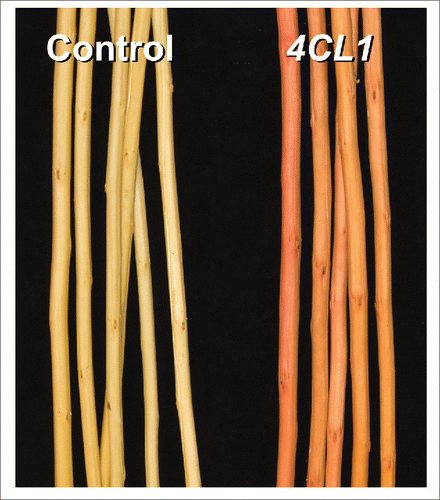
The first application of CRISPR/Cas9 genome editing in Populus targeted the 4-coumarate:CoA ligase (4CL) gene family involved in phenylpropanoid metabolism (Zhou et al., Citation2015). One of the genes, 4CL1 (Potri.001G036900), has been extensively characterized for its involvement in lignin biosynthesis. Down-regulation of 4CL1 or its orthologs leads to reduced lignin content and altered lignin structure in a number of species (Boerjan et al., Citation2003). Thus, CRISPR/Cas9 editing of 4CL1 served as a proof-of-concept study, allowing an assessment of its efficacy in perturbing lignin biosynthesis relative to previous methods. We generated 36 independent transgenic lines, and amplicon-sequencing of randomly selected events confirmed biallelic mutations in all cases (Zhou et al., Citation2015). As frequently reported for CRISPR/Cas9 genome editing, small indels, especially 1-bp modifications, were the predominant patterns and predicted to disrupt the reading frame. Consistent with this prediction, lignin content was reduced by ∼23% with a concomitant decrease of S/G lignin ratio by ∼30% in all transgenic plants (Zhou et al., Citation2015). Most telltale was the reddish-brown wood () observed in every single 4CL1 transgenic line. Wood discoloration is a known side effect of lignin modification frequently observed in transgenic plants with suppressed lignin gene expression. However, the coloration patterns reported in those cases lack the uniformity we observed for the CRISPR/Cas9 mutants. For example, antisense downregulation of 4CL1 in Populus resulted in patchy wood discoloration in only 5 of 14 transgenic lines (Voelker et al., Citation2010). In another case, wood discoloration patterns of transgenic Populus with down-regulated expression of cinnamoyl-CoA reductase were highly variable among vegetatively propagated plants (Van Acker et al., Citation2014), presumably due to the unstable nature of sense- or antisense-mediated post-transcriptional gene silencing. RNAi-mediated gene suppression is considered a more effective alternative, with improved trait stability (Li et al., Citation2008). However, huge variation in gene silencing efficiency remains. For instance, RNAi-silencing of 4-coumaroyl-CoA 3′-hydroxylase in transgenic Populus resulted in lignin reductions among 9 independent events that ranged from no change to 15–60% (Coleman et al., Citation2008). Similarly, RNAi silencing of 4CL in Pinus led to wood discoloration in only 2 of 12 viable transgenic lines (Wagner et al., Citation2009). By contrast, the homogeneity of wood discoloration both within and across independent CRISPR/Cas9-edited Populus mutant lines is consistent with a null 4CL1, and resembles that of naturally occurring “brown midrib” mutants of maize and sorghum (Sattler et al., Citation2010). The results serve as testament to the superiority of CRISPR/Cas9 over previous gene silencing methods.
Figure 1. Reddish-brown wood discoloration of 4CL1-edited Populus lines. The photo was modified from Zhou et al. Citation(2015) with permission.
A second gene, 4CL2 (Potri.019G049500) that is phylogenetically distinct from lignin-associated 4CLs (Chen et al., Citation2014). was also targeted in the study. 4CL2 has long been suggested to participate in flavonoid biosynthesis, based solely on its preferential expression in epidermis and roots (Harding et al., Citation2002). CRISPR/Cas9 mutation of 4CL2 was therefore expected to yield functional evidence to substantiate this role. As with the 4CL1 case, biallelic mutations were observed in all independent transgenic lines surveyed by amplicon-sequencing (Zhou et al., Citation2015). The metabolic consequence was examined in roots where flavonoid-derived condensed tannins (CTs) are known to accumulate to high levels (∼20% dry weight) in the experimental poplar genotype (Chen et al., Citation2014). CRISPR/Cas9 mutations of 4CL2 resulted in significantly reduced levels of condensed tannins in roots, providing reverse genetics evidence in support of Class II 4CL function in flavonoid biosynthesis.
After our initial publication, successful CRISPR/Cas9 editing was reported for Populus tomentosa (Fan et al., Citation2015). That study targeted a phytoene desaturase (PDS) gene for mutagenesis by multiplexing 4 gRNAs, and observed a ∼50% frequency of albino phenotypes. The lower mutation rates compared to our study may be attributed to gene redundancy and sequence heterozygosity that were not accounted for during gRNA design (see discussion below). Regardless, selected albino transgenic lines were sequence-confirmed to harbor biallelic mutations (Fan et al., Citation2015), providing additional support for the superior phenotypic uniformity of CRISPR/Cas9 mutants.
PRECISION EDITING BY CRISPR/CAS9
A major concern in genome editing is the specificity of the DNA modification, as off-target cleavage due to non-specific CRISPR/Cas9 activity may cause unintended mutations. This is especially true for plant genomes that are characterized by recurring episodes of whole-genome, segmental and tandem duplications. We assessed CRISPR/Cas9 specificity in Populus by analyzing its on-target versus off-target activities among duplicated genes. The gRNA for 4CL1 was designed to discriminate against the paralogous 4CL5 (Potri.003G188500), with 3 mismatches in the target sequence and one mismatch in the protospacer adjacent motif (PAM) (Zhou et al., Citation2015). By designing consensus primers flanking the target site, we were able to sequence both 4CL1 and 4CL5 amplicons simultaneously to assess the specificity of CRISPR/Cas9. A custom program, AGEseq (Analysis of Genome Editing by sequencing), was developed to facilitate variant detection (Xue and Tsai, Citation2015). No off-target cleavage in the 4CL5 locus was detected in 4CL1-edited lines, suggesting that CRISPR/Cas9 editing in Populus is highly specific (Zhou et al., Citation2015). The specificity was further demonstrated by a “negative” experiment, where mismatches between a 4CL5-gRNA, designed based on the P. trichocarpa reference genome (Phytozome v3), and the corresponding sequence of P. tremula x alba 717 used for transformation abolished cleavage in all transgenic lines examined (Zhou et al., Citation2015). In this case, the mismatches were in the form of 2 single nucleotide polymorphisms (SNPs), one per allele, located near or in the PAM. These results, together with the highly heterologous nature of woody perennials, suggest that off-target cleavage of CRISPR/Cas9 is likely to be low in these species.
The ability of CRISPR/Cas9 to discriminate single-base differences makes it a powerful tool to investigate functional redundancy of highly homologous genes that are otherwise difficult to discern using gene silencing approaches. For instance, 4CL1 is the most abundantly expressed isoform in Populus xylem, constituting >85% of 4CL transcripts, with 4CL5 being a distant second, based on RNA-Seq data (Swamy et al., Citation2015). However, the CRISPR/Cas9 knockout of 4CL1 reduced lignin content only by ∼23% (Zhou et al., Citation2015). Because the 4CL5 locus was unmodified, it is likely involved in the residual lignin biosynthesis of the 4CL1-knockout mutants. In the Populus study that targeted PDS, 4 gRNAs were designed based on one gene model (Potri.014G148700) without considering the genome duplicate (Potri.002G235200) that shares a high degree (93%) of coding sequence identity. While the genome sequence of P. tomentosa is not yet available, analysis of the P. trichocarpa and P. tremula x alba orthologs revealed different degrees of sequence variation between the 2 duplicates in the gRNA target regions. These differences (plus unknown allelic heterozygosity) likely contributed to the variation in the reported editing efficiencies of different gRNAs (Fan et al., Citation2015).
Several strategies are available to address functional redundancy. Examples include designing multigene-targeting gRNAs based on conserved sequences of homologous genes (Brooks et al., Citation2014; Jacobs et al., Citation2015), or multiplexing individual gRNA cassettes (Brooks et al., Citation2014; Fan et al., Citation2015; Zhang et al., Citation2014) for simultaneous editing of multiple genes. A recent study hijacked the tRNA processing machinery for efficient processing of up to 8, and potentially more, gRNAs from one single construct (Xie et al., Citation2015). In this design, the gRNA is positioned downstream of tRNA as a polycistronic gene with repeating tRNA-gRNA units for multiplexing (Xie et al., Citation2015). Because mature gRNAs are released following cleavage of the polycistronic transcript by the tRNA processing machinery, there is no sequence constraint at the 5′ end of gRNAs (Xie et al., Citation2015). This greatly simplifies gRNA design, as any sequences upstream of PAM (NGG) can be used (e.g., N20-NGG or other truncated lengths (Fu et al., Citation2014)), unlike with the standard design where gRNA is transcribed directly from the Pol III U6 or U3 promoter using a specific transcription initiation sequence (e.g., GN19-NGG or AN19-NGG, respectively) (Wang et al., Citation2008). In some cases, gRNA stacking, either by re-transformation or via controlled crosses of primary transformants may be necessary in order to discern individual gene functions. Combinatory approaches will increasingly be relied upon to investigate functional redundancy or complex pathways.
HARNESSING SEQUENCE POLYMORPHISMS OF OUTCROSSING SPECIES FOR EFFICIENT CRISPR/CAS9 EDITING
As discussed above, numerous studies have exploited sequence polymorphisms for developing gene- or allele-specific gRNAs to achieve precision editing by CRISPR/Cas9. However, few studies have addressed the added complexity of sequence polymorphisms on CRISPR/Cas9 genome editing, probably because the common plant models, such as Arabidopsis, rice, soybean and tomato, are selfing species with homozygous genomes. We argue that sequence polymorphisms are of concern in genome editing of most flowering plants, especially woody perennials that are outcrossing in nature. This is supported by the high frequency of genic SNPs reported for trees, one per 60 bp in Populus tremula (Ingvarsson, Citation2005) or one per 16-33 bp in Eucalyptus depending on the species (Külheim et al., Citation2009). A recent population genomics study of 544 Populus trichocarpa individuals identified 17.9 million (M) SNPs by whole-genome resequencing (Evans et al., Citation2014). Using a similar approach with both resequencing and RNA-Seq data, we uncovered over 10 M SNPs from a single individual, P. tremula x alba clone 717 that is commonly used in Populus transformation (Xue et al., Citation2015). Given the high levels of heterozygosity in tree species, the need to consider sequence polymorphisms in genome editing experiments cannot be over-emphasized.
Current genome data mining tools and resources do not support exploration of sequence variations. In fact, most published genome sequences are, by default, consensus sequences of their (pseudo)haploid genomes. Thus, while several web-based gRNA design programs are available for plants, such as CRISPR-PLANT (Xie et al., Citation2014). and CRISPR-P (Lei et al., Citation2014), their utility for outcrossing species is limited because the preloaded genome sequences lack biallelic (or multiploid) coverage. Furthermore, it is often the case as in Populus that the species or genotype chosen for whole genome sequencing (e.g.,, P. trichocarpa Nisqually-1) is different from that routinely used for genetic transformation (e.g., P. tremula x alba 717). To facilitate CRISPR/Cas9 genome editing in 717, we constructed a custom 717 genome by substituting >10 M sequence variants into the P. trichocarpa reference genome (Xue et al., Citation2015). We then assessed the impact of sequence polymorphisms on gRNA specificity for 1000 randomly selected genes. gRNAs were designed based on the haploid genome of (1) P. trichocarpa or (2) 717, and then cross-checked for SNPs or indels in the 717 target sequences. From the first analysis, we found that more than 57% of the gRNAs designed based on the P. trichocarpa genome correspond to regions that harbor SNPs or indels in 717 (Xue et al., Citation2015). These sequence variations can be attributed to genotypic as well as allelic differences. The rates were slightly lower but nevertheless high (42-44%) in the second analysis when the custom 717 genome was used, owing to allelic variations (Zhou et al., Citation2015). Together, these data suggest that standard gRNA designs may suffer from a high probability of failure in CRISPR/Cas9 genome editing, due to the frequent occurrence of SNPs/indels in outcrossers. The 717 sequences and associated BLAST and gene model query tools are available via AspenDB (http://aspendb.uga.edu/s717). The “Probe Search” tool is particularly useful for screening custom gRNAs or PCR primers against known SNPs/indels in the P. trichocarpa or the custom 717 genome (Xue et al., Citation2015). A set of pre-selected gRNAs with no known sequence variants for 38,509 Populus genes is provided (Zhou et al., Citation2015). The modified, variant-sensitive pipeline for gRNA design, called “Aspen CRISPR designer,” is also available at AspenDB and can be applied to other species.
For outcrossing species lacking a genomic variant database, several approaches can be applied to identify SNPs/indels in the gene(s) of interest prior to gRNA selection. RNA-Seq data from the transformation genotype, when available, is an excellent source for high-confidence SNP discovery. Provided that the gene of interest is reasonably expressed (e.g.,, with a high read coverage), manual inspection of candidate gRNAs against RNA-Seq alignments using programs such as the popular Integrative Genomics Viewer (http://www.broadinstitute.org/igv) is usually effective to confirm their specificity. In the absence of deep-sequencing data, PCR primers (with or without degeneracy) can be designed to amplify the target gene coding region based on a closely-related reference genome, or on conserved amino acid sequences of orthologs as in conventional RT-PCR cloning (Hu et al., Citation1998). As PAM (NGG)–the only sequence constraint in gRNA design—occurs frequently, it is not necessary to obtain full-length gene sequences. However, the use of multiple primer pairs is recommended to increase the likelihood of capturing both alleles. Direct sequencing of PCR products to identify sequence variants is an established practice in molecular ecology research, and several programs have been developed to decode overlapping Sanger chromatograms (Dmitriev and Rakitov, Citation2008). For researchers with convenient access to high-throughput sequencing, barcoded amplicons can be sequenced as spike-in's. Sequence reads can then be processed by AGEseq to search for potential variants in the target gene sequences (Xue and Tsai, Citation2015). AGEseq is available as a standalone program or a Galaxy-compatible tool (https://toolshed.g2.bx.psu.edu) to support web-based data analysis. It should be noted that PCR amplification of noncoding sequences can be tricky due to the lower degree of sequence conservation (Xue et al., Citation2015). Thus, for CRISPR/Cas9 applications that target promoter or other noncoding sequences (e.g., transcriptional activation or chromatin modification (Sander and Joung, Citation2014)), resequencing of the transformation clone for unbiased variant calling, as discussed above for Populus 717 (Xue et al., Citation2015), is recommended.
CONCLUDING REMARKS AND FUTURE PROSPECTS
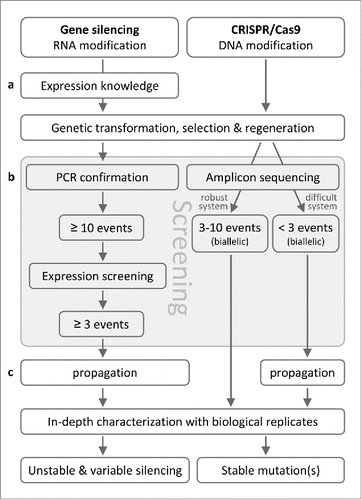
The Populus study extended the ever growing list of plant species successfully genome-edited by CRISPR/Cas9 (Bortesi and Fischer, Citation2015; Kumar and Jain, Citation2015) to woody perennials. The significance for biotechnological improvement is several fold (). Woody perennials suffer as experimental systems due to the lengthiness of genetic transformation and plant characterization. Many forest and horticultural tree species and genotypes are notoriously recalcitrant to tissue culture regeneration with low transformation efficiencies (Litz and Padilla, Citation2012; Busov et al., Citation2005). Limitations associated with other established technologies, such as incomplete silencing, position effects or epigenetic modifications of transgenes (Matzke and Matzke, Citation1998), further impede progress in transgenic tree research. Even for species with a facile transformation system, such as Populus tremula x alba 717, the common practice of generating multiple independent events for preliminary screening prior to in-depth characterization of selected lines is resource-demanding and time-consuming (). The ability of CRISPR/Cas9 to generate null mutations in primary transformants, and the promise of phenotypic consistency among independent biallelic mutants as demonstrated in the Populus study make a compelling argument that it may soon become acceptable to characterize only a few transgenic lines with sequence-verified mutations. High-throughput methods for mapping genome editing patterns (Xue and Tsai, Citation2015) and transgene (e.g., T-DNA harboring Cas9, gRNA and selectable marker) insertion sites (Kanizay et al., Citation2015) are expected to facilitate this effort. As depicted in , primary transformants with confirmed editing can be used directly for in-depth characterization when multiple independent events are available as biological replicates (Zhou et al., Citation2015). Even when only a few mutation events are obtained (for difficult-to-transform species or from suboptimal transformation trials), the ability to bypass RNA-level screening and to obtain stable mutations still confers advantages over the gene-silencing approaches. CRISPR/Cas9 thus offers the enticing prospect of streamlined transgenic characterization and expedited large-scale functional characterization.
Figure 2. Schematic comparisons of experimental approaches involving gene silencing vs. CRISPR/Cas9 genome editing. (a) For gene silencing by antisense, sense or RNAi approaches, knowledge about the spatiotemporal expression pattern of the target gene is used to select an appropriate promoter for construct preparation. Expression knowledge is not necessary for CRISPR/Cas9 editing at the DNA level, and vectors with a Pol III promoter (e.g., U6 or U3) for gRNA expression and a Cas9 under control of a constitutive (e.g., 35S) promoter are widely applicable. (b) Following regeneration of putative transgenic plants, conventional screening involves PCR confirmation of transgenes followed by expression analysis to select events with desired (or maximum) levels of gene suppression. It is common to screen a large number of events (≥10) to identify a minimum of 3 for subsequent analysis. This process can be greatly simplified with CRISPR/Cas9 mutants, as DNA level analysis by amplicon sequencing can identify events with biallelic mutations for further analysis. No RNA level analysis is necessary. (c) For species with a robust transformation system, multiple biallelic CRISPR/Cas9 events can be used directly as biological replicates for in-depth characterization. In case of difficult-to-transform species for which few biallelic events may be obtained, micropropagation is necessary to obtain biological replicates before in-depth characterization, as is typically done using gene silencing.
CRISPR/Cas9 also promises to accelerate genetic improvement of woody perennials. Biallelic mutations of the target gene(s) in conjunction with the hemizygous nature of transgene (e.g., T-DNA) integration in primary transformants (T0) means that DNA-edited but transgene-free progenies can be readily obtained from T1 segregants, as demonstrated in annual species (Zhang et al., Citation2014; Zhou et al., Citation2014). A major limitation is the long juvenile phase of most woody species. Trait stacking with early flowering induction is thus highly desirable, for example by over-expression of Flowering Locus T (FT) as demonstrated in Prunus (plum), Populus and Eucalyptus (Hoenicka et al., Citation2014; Klocko et al., Citation2015; Srinivasan et al., Citation2012). Another concern for outcrossing species is that the progenies will be different from the parents, which necessitates additional screening and selection. We note, however, that introgressing transgenic/mutant events into elite genotypes is an established practice in crop and tree breeding programs, and hence not unique to CRISPR/Cas9-edited mutants. A well-known tree example is the loblolly pine (Pinus taeda) clone 7–56 that harbors a naturally occurring null allele of cinnamoyl alcohol dehydrogenase involved in lignin biosynthesis, and that is extensively used as a parent in several breeding programs in the US (Gill et al., Citation2003). Another example is early-flowering FT-plum that serves as a parent in the FastTrack breeding program to expedite development of improved commercial cultivars (Callahan et al., Citation2015). Various molecular and genomics tools are available to facilitate progeny selection in transgenic breeding programs. Recently, virus-based systems have been explored for transient expression of genome-engineering reagents (Ali et al., Citation2015; Baltes et al., Citation2014). However, such systems still utilize T-DNA to express the repurposed viral genome components, and the resulting plants are not transgene-free without crossing. These methods also depend on agroinfiltration to deliver the vector into plant cells, which unfortunately is not applicable to most woody species with an open minor vein structure in their leaves (Gamalei, Citation1989). Clearly, development of innovative methods will be necessary for facile removal of T-DNA from the genome-edited plants without crossing. Regardless, null segregants of crop cultivars derived from genome editing are indistinguishable from naturally occurring or chemically-induced mutants, and may therefore be exempt from GMO regulation in countries that adhere to product-based regulations (Voytas and Gao, Citation2014). This is expected to significantly reduce the timeline and financial burden associated with developing new crop varieties, thereby encouraging adoption of CRISPR/Cas9-based genome editing for the forest products, fruit/tree nuts, and woody ornamental industries.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
Funding
The research in the Tsai laboratory discussed in this article was supported by the Department of Energy, Office of Biological and Environmental Research (grant no. DE-SC0008470), the Department of Agriculture, National Institute of Food and Agriculture (grant no. 2015-67013-22812), and by the Georgia Research Alliance-Hank Haynes Forest Biotechnology endowment.
REFERENCES
- Ali Z, Abul-faraj A, Li L, Ghosh N, Piatek M, Mahjoub A, Aouida M, Piatek A, Baltes Nicholas J, Voytas Daniel F, et al. Efficient virus-mediated genome editing in plants using the CRISPR/Cas9 system. Molecular Plant 2015; 8:1288-91; PMID:25749112; http://dx.doi.org/10.1016/j.molp.2015.02.011
- Baltes NJ, Gil-Humanes J, Cermak T, Atkins PA, Voytas DF. DNA replicons for plant genome engineering. Plant Cell 2014; 26:151-63; PMID:24443519; http://dx.doi.org/10.1105/tpc.113.119792
- Boerjan W, Ralph J, Baucher M. Lignin biosynthesis. Annu Rev Plant Biol 2003; 54:519-46; PMID:14503002; http://dx.doi.org/10.1146/annurev.arplant.54.031902.134938
- Bortesi L, Fischer R. The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol Adv 2015; 33:41-52; PMID:25536441; http://dx.doi.org/10.1016/j.biotechadv.2014.12.006
- Brooks C, Nekrasov V, Lippman ZB, Van Eck J. Efficient gene editing in tomato in the first generation using the clustered regularly interspaced short palindromic repeats/CRISPR-associated9 system. Plant Physiol 2014; 166:1292-7; PMID:25225186; http://dx.doi.org/10.1104/pp.114.247577
- Busov VB, Brunner AM, Meilan R, Filichkin S, Ganio L, Gandhi S, Strauss SH. Genetic transformation: a powerful tool for dissection of adaptive traits in trees. New Phytologist 2005; 167:9-18; PMID:15948825; http://dx.doi.org/10.1111/j.1469-8137.2005.01412.x
- Callahan A, Dardick C, Tosetti R, Lalli D, Scorza R. 21st Century Approach to Improving Burbank's ‘Stoneless’ Plum. HortScience 2015; 50:195-200
- Carroll D. Genome engineering with targetable nucleases. Annu Rev Biochem 2014; 83:409-39; PMID:24606144; http://dx.doi.org/10.1146/annurev-biochem-060713-035418
- Chen H-Y, Babst BA, Nyamdari B, Hu H, Sykes R, Davis MF, Harding SA, Tsai C-J. Ectopic expression of a loblolly pine Class II 4-coumarate:CoA ligase alters soluble phenylpropanoid metabolism but not lignin biosynthesis in Populus. Plane Cell Physiol 2014; 55:1669-78; http://dx.doi.org/10.1093/pcp/pcu098
- Coleman HD, Park JY, Nair R, Chapple C, Mansfield SD. RNAi-mediated suppression of p-coumaroyl-CoA 3′-hydroxylase in hybrid poplar impacts lignin deposition and soluble secondary metabolism. Proc Natl Acad Sci 2008; 105:4501-6; http://dx.doi.org/10.1073/pnas.0706537105
- Dmitriev DA, Rakitov RA. Decoding of superimposed traces produced by direct sequencing of heterozygous indels. PLoS Comput Biol 2008; 4:e1000113; PMID:18654614
- Evans LM, Slavov GT, Rodgers-Melnick E, Martin J, Ranjan P, Muchero W, Brunner AM, Schackwitz W, Gunter L, Chen JG, et al. Population genomics of Populus trichocarpa identifies signatures of selection and adaptive trait associations. Nat Genet 2014; 46:1089-96; PMID:25151358; http://dx.doi.org/10.1038/ng.3075
- Fan D, Liu T, Li C, Jiao B, Li S, Hou Y, Luo K. Efficient CRISPR/Cas9-mediated targeted mutagenesis in Populus in the first generation. Scientific Reports 2015; 5:12217; PMID:26193631; http://dx.doi.org/10.1038/srep12217
- Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotech 2014; 32:279-84; http://dx.doi.org/10.1038/nbt.2808
- Gamalei Y. Structure and function of leaf minor veins in trees and herbs. Trees 1989; 3:96-110; http://dx.doi.org/10.1007/BF01021073
- Gill GP, Brown GR, Neale DB. A sequence mutation in the cinnamyl alcohol dehydrogenase gene associated with altered lignification in loblolly pine. Plant Biotechnol J 2003; 1:253-8; PMID:17163902; http://dx.doi.org/10.1046/j.1467-7652.2003.00024.x
- Harding SA, Leshkevich J, Chiang VL, Tsai CJ. Differential substrate inhibition couples kinetically distinct 4-coumarate:coenzyme A ligases with spatially distinct metabolic roles in quaking aspen. Plant Physiol 2002; 128:428-38; PMID:11842147; http://dx.doi.org/10.1104/pp.010603
- Hoenicka H, Lehnhardt D, Nilsson O, Hanelt D, Fladung M. Successful crossings with early flowering transgenic poplar: interspecific crossings, but not transgenesis, promoted aberrant phenotypes in offspring. Plant Biotechnol J 2014; 12:1066-74; PMID:24975279; http://dx.doi.org/10.1111/pbi.12213
- Hu W-J, Kawaoka A, Tsai CJ, Lung J, Osakabe K, Ebinuma H, Chiang VL. Compartmentalized expression of two structurally and functionally distinct 4-coumarate:CoA ligase genes in aspen (Populus tremuloides). Proc Natl Acad Sci USA 1998; 95:5407-12; PMID:9560289; http://dx.doi.org/10.1073/pnas.95.9.5407
- Ingvarsson PK. Nucleotide polymorphism and linkage disequilibrium within and among natural populations of European aspen (Populus tremula L., Salicaceae). Genetics 2005; 169:945-53; PMID:15489521; http://dx.doi.org/10.1534/genetics.104.034959
- Jacobs TB, LaFayette PR, Schmitz RJ, Parrott WA. Targeted genome modifications in soybean with CRISPR/Cas9. BMC Biotechnol 2015; 15:16; PMID:25879861; http://dx.doi.org/10.1186/s12896-015-0131-2
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012; 337:816-21; PMID:22745249; http://dx.doi.org/10.1126/science.1225829
- Kanizay LB, Jacobs TB, Gillespie K, Newsome JA, Spaid BN, Parrott WA. HtStuf: High-throughput sequencing to locate unknown DNA junction fragments. Plant Genome 2015; 8(1); http://dx.doi.org/10.3835/plantgenome2014.10.0070
- Klocko AL, Ma C, Robertson S, Esfandiari E, Nilsson O, Strauss SH. FT overexpression induces precocious flowering and normal reproductive development in Eucalyptus. Plant Biotechnol J 2015; PMID:26132805
- Külheim C, Hui Yeoh S, Maintz J, Foley WJ, Moran GF. Comparative SNP diversity among four Eucalyptus species for genes from secondary metabolite biosynthetic pathways. BMC Genomics 2009; 10:452; PMID:19775472; http://dx.doi.org/10.1186/1471-2164-10-452
- Kumar V, Jain M. The CRISPR-Cas system for plant genome editing: advances and opportunities. J Exp Botany 2015; 66:47-57; PMID:25371501; http://dx.doi.org/10.1093/jxb/eru429
- Lei Y, Lu L, Liu HY, Li S, Xing F, Chen L-L. CRISPR-P: A web tool for synthetic single-guide RNA design of CRISPR-system in plants. Molecular Plant 2014; 7:1494-6; PMID:24719468; http://dx.doi.org/10.1093/mp/ssu044
- Li J, Brunner A, Shevchenko O, Meilan R, Ma C, Skinner J, Strauss S. Efficient and stable transgene suppression via RNAi in field-grown poplars. Transgenic Res 2008; 17:679-94; PMID:17929189; http://dx.doi.org/10.1007/s11248-007-9148-1
- Litz R, Padilla G. Genetic transformation of fruit trees. In: Schnell RJ, Priyadarshan PM, eds., Genomics of Tree Crops. New York: Springer; 2012:117-53
- Matzke AJM, Matzke MA. Position effects and epigenetic silencing of plant transgenes. Curr Opinion Plant Biol 1998; 1:142-8; PMID:10066569; http://dx.doi.org/10.1016/S1369-5266(98)80016-2
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotech 2014; 32:347-55; http://dx.doi.org/10.1038/nbt.2842
- Sattler SE, Funnell-Harris DL, Pedersen JF. Brown midrib mutations and their importance to the utilization of maize, sorghum, and pearl millet lignocellulosic tissues. Plant Sci 2010; 178:229-38; http://dx.doi.org/10.1016/j.plantsci.2010.01.001
- Srinivasan C, Dardick C, Callahan A, Scorza R. Plum (Prunus domestica) trees transformed with poplar FT1 result in altered architecture, dormancy requirement, and continuous flowering. PLoS ONE 2012; 7:e40715; PMID:22859952; http://dx.doi.org/10.1371/journal.pone.0040715
- Swamy PS, Hu H, Pattathil S, Maloney VJ, Xiao H, Xue LJX, Chung JD, Johnson VE, Zhu Y, Peter G, et al. Tubulin perturbation leads to unexpected cell wall modifications and affects leaf expansion and stomatal behavior in Populus. J Exp Botany 2015; http://dx.doi.org/10.1093/jxb/erv383
- Van Acker R, Leplé JC, Aerts D, Storme V, Goeminne G, Ivens B, Légée F, Lapierre C, Piens K, Van Montagu MCE, et al. Improved saccharification and ethanol yield from field-grown transgenic poplar deficient in cinnamoyl-CoA reductase. Proc Natl Acad Sci 2014; 111:845-50; http://dx.doi.org/10.1073/pnas.1321673111
- Voelker SL, Lachenbruch B, Meinzer FC, Jourdes M, Ki C, Patten AM, Davin LB, Lewis NG, Tuskan GA, Gunter L, et al. Antisense down-regulation of 4CL expression alters lignification, tree growth, and saccharification potential of field-grown poplar. Plant Physiol 2010; 154:874-86; PMID:20729393; http://dx.doi.org/10.1104/pp.110.159269
- Voytas DF, Gao C. Precision genome engineering and agriculture: Opportunities and regulatory challenges. PLoS Biol 2014; 12:e1001877; PMID:24915127; http://dx.doi.org/10.1371/journal.pbio.1001877
- Wagner A, Donaldson L, Kim H, Phillips L, Flint H, Steward D, Torr K, Koch G, Schmitt U, Ralph J. Suppression of 4-Coumarate-CoA Ligase in the Coniferous Gymnosperm Pinus radiata. Plant Physiol 2009; 149:370-83; PMID:18971431; http://dx.doi.org/10.1104/pp.108.125765
- Wang MB, Helliwell CA, Wu LM, Waterhouse PM, Peacock WJ, Dennis ES. Hairpin RNAs derived from RNA polymerase II and polymerase III promoter-directed transgenes are processed differently in plants. RNA 2008; 14:903-13; PMID:18367720; http://dx.doi.org/10.1261/rna.760908
- Xie K, Minkenberg B, Yang Y. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc Natl Acad Sci 2015; 112:3570-5; http://dx.doi.org/10.1073/pnas.1420294112
- Xie K, Zhang J, Yang Y. Genome-wide prediction of highly specific guide RNA spacers for CRISPR–Cas9-mediated genome editing in model plants and major crops. Molecular Plant 2014; 7:923-6; PMID: 24482433; http://dx.doi.org/10.1093/mp/ssu009
- Xue L-J, Alabady MS, Mohebbi M, Tsai CJ. Exploiting genome variation to improve next-generation sequencing data analysis and genome editing efficiency in Populus tremula x alba 717-1B4. Tree Genetics Genomics 2015; 11:82; http://dx.doi.org/10.1007/s11295-015-0907-5
- Xue LJ, Tsai CJ. AGEseq: Analysis of genome editing by sequencing. Mol Plant 2015; 8:1428-1430; http://dx.doi.org/10.1016/j.molp.2015.06.001
- Zhang H, Zhang J, Wei P, Zhang B, Gou F, Feng Z, Mao Y, Yang L, Zhang H, Xu N, et al. The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnology J 2014; 12:797-807; PMID:24854982; http://dx.doi.org/10.1111/pbi.12200
- Zhou H, Liu B, Weeks DP, Spalding MH, Yang B. Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res 2014; 42:10903-14; PMID:25200087; http://dx.doi.org/10.1093/nar/gku806
- Zhou X, Jacobs TB, Xue LJ, Harding SA, Tsai CJ. Exploiting SNPs for biallelic CRISPR mutations in the outcrossing woody perennial Populus reveals 4-coumarate: CoA ligase specificity and redundancy. New Phytol 2015; 208:298-301; http://dx.doi.org/10.1111/nph.13470