
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The MAB phases are a family of layered ternary transition metal borides, with atomically laminated crystal structures comprised of transition metal boride (M-B) layers interleaved by single, or double, Al (A) layers. Herein, density functional theory is implemented to evaluate the thermodynamic stability of disordered (Mn1-xCrx)2AlB2, and disordered and ordered (Mn1-xCrx)3AlB4 quaternaries. The (Mn1-xCrx)2AlB2 solid solutions were synthesized over the entire range of substitution. A (Mn1-xCrx)3AlB4 solid solution was produced, on the base of Cr3AlB4, to form (Mn0.33Cr0.66)3AlB4. Powder X-ray diffraction shows lattice parameter shifts and unit cell expansions indicative of successful solid solution formations.
GRAPHICAL ABSTRACT
IMPACT STATEMENT
In this work, first-principles calculations evaluate the stability of (Mn1-xCrx)2AlB2 and (Mn1-xCrx)3AlB4 solid solutions. Experimentally, MAB phase solid solutions of (Mn1-xCrx)2AlB2 and (Mn0.33Cr0.66)3AlB4 are synthesized for the first time.
Introduction
The MAB phases are a family of atomically laminated, ternary, transition metal borides, wherein M is a transition metal, A is generally aluminium, Al, and B is boron. The first MAB phase, MoAlB, was synthesized in 1966 by Jeitschko et al. [Citation1], followed by Mn2AlB2 [Citation2] and Fe2AlB2 [Citation3] later that same year. The ternaries, Cr2AlB2 and Cr3AlB4 were not synthesized until 1973 by Chaban and Kuz’ma [Citation4]. Recently, there has been a renewed interest in these phases due to their unique magneto-caloric properties [Citation5], high-temperature oxidation resistance [Citation6] and their role as a precursor for 2-D, MXene-like borides [Citation7,Citation8]. A more complete history and summary of the MAB phases and their properties can be found in Ref. [Citation9].
The MAB phase structure contains either single, or double, Al atomic layers sandwiched between two M-B layers (Figure (a–b)) [Citation10]. All structures examined herein contain a single Al atomic layer. The 212 MABs, Mn2AlB2 and Cr2AlB2, are isostructural and crystallize in the Cmmm space group [Citation11]. To date the only 314 MAB phase synthesized is Cr3AlB4 that crystallizes in the Pmmm space group. Density functional theory, DFT, calculations have shown that other 314 MAB phases, such as Mn3AlB4 and Fe3AlB4, to be thermodynamically unstable vis-à-vis other competing phases [Citation12]. In the 212 MAB structures, the layers stack in the b-direction; in the 314 they stack in the c-direction.
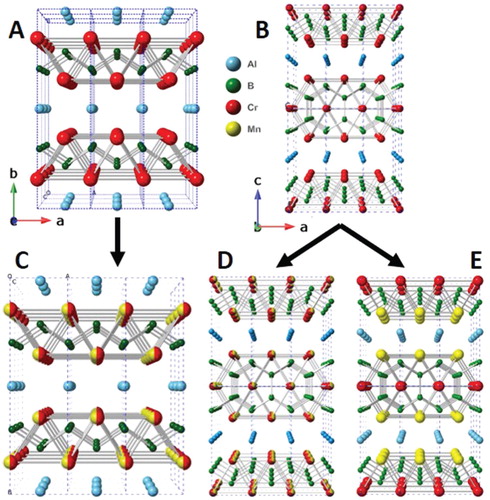
Figure 1. Crystal structures of, a) M2AlB2, b) M3AlB4, c) disordered (Mn1-xCrx)2AlB2, d) disordered (Mn1-xCrx)3AlB4 and e) ordered Mn2CrAlB4. Note that in the 212 structure (a), the layers stack along the b-direction, while in the 314 (b) they stack along the c-direction.
Solid solutions on the M sites allow for tuning of material properties as a function of substitution. In the MAB phases, partial M-site substitutions have been successful for MoAlB, WAlB and Fe2AlB2. In 1995, Yu et al. were the first to synthesize single crystals of the solid solutio, (Mo0.61Cr0.39)AlB [Citation13]. Okada et al. later expanded the doping range, as well as synthesizing a (Mo1-xWx)AlB quaternary [Citation14]. Increasing the W content led to an increase in hardness, while increasing the Cr substitution lowered the resistivity of the material. Solid solutions of (Fe1-xM′x)2AlB2 have been synthesized wherein M′ = Mn [Citation15] or Co [Citation16] with substitutions up to x = 1 and x = 0.15, respectively. The solid solutions were shown to increase the magneto-caloric effect over a greater range of temperatures. DFT calculations suggest that ordered (M1-xM′x)3AlB4 compounds may be stable. However, since these calculations do not take into account the formation of competing ordered 212 phases, these conclusions, as shown below, are incorrect [Citation17].
The purpose of this work was to expand the range of MAB solid solutions in the Mn-Cr-Al-B system. This endeavour has been fruitful in that we discovered that solid solutions in (Mn1-xCrx)2AlB2 exist for all x. We also discovered the existence of a (Mn1-xCrx)3AlB4 solid solution for x = 0.33.
Methods
Computational
All first-principles calculations were performed by means of DFT and the projector augmented wave method [Citation18,Citation19], as implemented within the Vienna ab-initio simulation package (VASP) version 5.4.1 [Citation20,Citation21,Citation22], with a plane-wave energy cut-off of 400 eV. The spin-polarized version of the generalized gradient approximation (GGA), as parameterized by Perdew–Burke–Ernzerhof (PBE) [Citation23], was used for treating the electron exchange and correlation effects. Multiple spin configurations were considered; the results presented are the ones for the lowest energy spin configuration. The Brillouin zone was integrated by Monkhorst–Pack special k-point sampling [Citation24]. The total energy was minimized through relaxation of unit-cell shape and volume, and internal atomic positions until satisfying an energy convergence of 10−5 eV/atom and force convergence of 10−2 eV/Å.
Chemical disorder of Mn and Cr in the (Mn1-xCrx)2AlB2 and (Mn1-xCrx)3AlB4 quaternaries were modelled on the M-sublattice using the special quasi-random structure (SQS) method [Citation25]. Convergency tests show that supercells with 120 or more atoms give a qualitatively accurate representation and a quantitative convergency in terms of calculated formation enthalpies, equilibrium volumes and lattice parameters.
The thermodynamic stabilities of (Mn1-xCrx)2AlB2 and (Mn1-xCrx)3AlB4 were investigated at 0 K with respect to their decomposition into any combination of competing phases. The set of most competing phases, denoted the equilibrium simplex, is identified using a linear optimization procedure [Citation26,Citation27]. The stability of a phase is quantified in terms of the formation enthalpy, ΔHcp, by comparing its energy to the energy of the equilibrium simplex according to
(1)
(1) A phase is concluded to be stable when ΔHcp < 0.
We approximate contributions from other temperature-dependent effects, e.g. lattice vibrations and electronic entropy, to the formation enthalpy to be negligible as such contributions from a phase, significant or not, tend to be cancelled out in the Gibbs free energy of formation terms [Citation28]. However, since we are investigating solid solutions of Mn and Cr, approximated through modelled disorder (SQS), the contributions from configurational entropy to the Gibbs free energy of formation , when T ≠ 0 K, are approximated by
(2)
(2) where the entropic contribution, ΔS, assuming an ideal solution of Mn and Cr on the M-site, is given by
(3)
(3) where kB is the Boltzmann constant and x is the concentration of Cr on the M-sublattice.
The structure of each of the three phases modelled can be visualized such that that the disordered structure is a mixture of Mn and Cr atoms on the M lattice sites (Figure (c–d)), while in the ordered Mn2CrAlB4, the M site, nearest to the Al layer is replaced by Mn (Figure (e)).
Experimental details
Samples were prepared via a powder metallurgical route starting with elemental Mn (Alfa Aesar, 99.6%, <10 µm), Cr (Alfa Aesar, 99.2%, <10 µm), Al (Alfa Aesar, 99.5%, <44 µm), CrB (Grade B, H.C. Starck, 0.1–0.3% Fe impurities), amorphous B (Ba) (Alfa Aesar, 98%, <44 µm) and crystalline B (Bc) (Alfa Aesar, 98%, <44 µm) powders. For the 212 solid solutions, the synthesis was carried out in a two-step method wherein Mn, Cr and Bc were first reacted to form (Mn1-xCrx)B solid solutions. This was followed by reaction of the latter with Al to form the corresponding MAB phase. To form the initial (Mn1-xCrx)B solid solutions, a total of 2.5 g of powders were mixed, where x = 0.10, 0.25, 0.50, 0.75 and 0.9. These molar ratios were selected to cover the entire solid solution range. For the 314 solid solutions, powders were mixed in molar ratios of (Mn1-xCrBx)3Al1.15B3(1-x)+1, where x = 0.33, 0.66 and 1. The powder mixtures were ball milled for 24 h in plastic jars using yttria-stabilized zirconia media. Green bodies were cold pressed at 300 MPa from the mixtures to form 13 mm diameter, ≈10 mm high cylinders. The (Mn1-xCrx)B pellets were reacted via pressureless sintering in a horizontal tube furnace, heated at a rate of 5°C/min and held at 1050°C for 5 h, before cooling to ambient temperatures at the same rate. The reaction was conducted in a flowing Ar atmosphere, with Ti powder upstream gettering oxygen. The reacted pellets were crushed to fine powders using an agate mortar and pestle, mixed with Al powder in the ratio of (Mn1-xCrx)2Al1.2B2, ball milled and cold-pressed in the same method as above. The pellets were sintered at 1050°C for 5 h in a horizontal tube furnace under the same conditions as above.
The two-step reaction sintering for the M3AlB4 sample was slightly different. First, the green bodies were reacted at 970°C for 1 h, then crushed and re-pressed; followed by a second sintering at 1050°C for 10 h.
After reaction, the samples were ground into fine powders using an agate mortar and pestle. X-ray diffraction (XRD) was performed using a diffractometer (Rigaku SmartLab) using a Cu Kα source. The step size and dwell times were 0.02° and 1.8 s/step, respectively. A graphite monochromator was utilized to remove background fluorescence. Small amounts of Si powder were added to the powders as an internal reference to more accurately determine the lattice parameters, LPs. XRD analysis was conducted using Jade 6 software. The quantitative analysis for the 314 phase was performed with the Easy Quant package using the RIR method [Citation29].
Results and discussion
Computational results
For the evaluation of the thermodynamic phase stability of (Mn1-xCrx)2AlB2 and (Mn1-xCrx)3AlB4, we compare their calculated energies to an identified set of most competing phases (equilibrium simplex) in terms of the formation enthalpy ΔHcp using Equation 1. The identified equilibrium simplex for each composition is given in Table . Figure (a) shows the calculated phase stabilities at 0 K (ΔHcp) and at elevated temperatures (ΔGcp). In this system, both Mn2AlB2 and Cr2AlB2 are stable with calculated formation enthalpies of −67 and −30 meV/atom, respectively. In Figure (a), these values are used as reference points, i.e. are set to zero. The (Mn1-xCrx)2AlB2 solid solutions, with disorder on the M sites, are predicted to be stable at all x (Figure (a)). At elevated temperatures, the entropy contribution due to the disorder further stabilizes the solids solutions with respect to decomposition into Mn2AlB2 and Cr2AlB2 (Figure (a)).
Figure 2. Calculated formation enthalpies, ΔH, at 0 K and Gibbs free energies of formation, ΔG, at T > 0 K for, a) (Mn1-xCrx)2AlB2 and, b) (Mn1-xCrx)3AlB4 solid solutions. In (a), enthalpies of ternaries, viz. −67 and −30 meV/atom for Mn2AlB2 and Cr2AlB2 respectively, are used as reference points (zero).
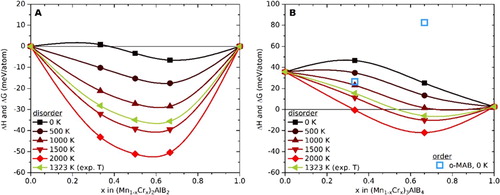
Table 1. Identified equilibrium simplex for (Mn1-xCrx)2AlB2 and (Mn1-xCrx)3AlB4 phases
Figure (b) shows the calculated stability for the (Mn1-xCrx)3AlB4 quaternaries Both disorder of Mn and Cr on the M-sublattices (solid symbols) as well as an out-plane chemically ordered structures (open squares), here referred to as o-MAB, were considered. At 0 K, only Cr3AlB4 is found to be close to stable with ΔH = +3 meV/atom. None of the considered ordered o-MAB structures, viz. (MnCr2)AlB4 nor (Mn2Cr)AlB4, were found to be stable. Disorder of the Mn and Cr atoms, however, are stabilized with increasing temperatures, but only if the solid solutions are Cr-rich (Figure (b)). At 1323 K (1050°C), the experimental condition used herein, (Mn0.33Cr0.66)3AlB is predicted to be stable, but (Mn0.66Cr0.33)3AlB4 is not.
The theoretical lattice parameters and unit cell volumes, Vuc, were calculated for both the (Mn1-xCrx)2AlB2 and (Mn1-xCrx)3AlB4 systems. As the entire (Mn1-xCrx)2AlB2 system was found to be stable in the disordered state, only this state was considered, with the calculated values presented in Table . For the (Mn1-xCrx)3AlB4 system, both disordered and ordered structures were evaluated, and the calculated values are listed in Table .
Table 2. Calculated and experimental LPs and Vuc of (Mn1-xCrx)2AlB2 solid solutions.
Table 3. Experimental and calculated LPs and Vuc for (Mn1-xCrx)3AlB4 with disorder (D) and order (O) of Mn and Cr.
Experimental results
(Mn1-xCrx)2AlB2
XRD patterns of the (Mn1-xCrx)2AlB2 solid solutions, reacted at 1050°C for 5 h, are shown in Figure (a). The XRD patterns for the Cr-rich solid solutions show residual CrB peaks that increase in intensity as the Cr content increases. As shown in previous work [Citation8,Citation30], Cr2AlB2 may require a second sintering, or excess Al, to fully react. As these compositions approach Cr2AlB2, the 20% excess Al is insufficient to react with all the CrB in a single reaction step, resulting in some impurity phases.
Figure 3. a) XRD patterns of (Mn1-xCrx)2AlB2 sold solutions reacted at 1050°C for 5 h as a function of x. Si powder was added as a reference. b) Lattice parameters, and c) unit cell volumes of (Mn1-xCrx)2AlB2 solid solutions as a function x. Solid lines and symbols denote the experimental LPs; dashed lines and open symbols, denote calculated LPs.
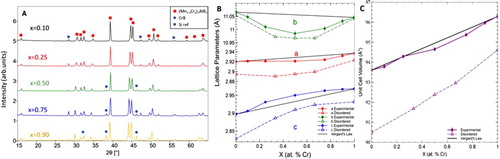
Figure 4. Structural characteristics of (Mn1-xCrx)3AlB4 solid solutions, a) XRD patterns of Cr3AlB4 and (Mn0.33Cr0.66)3AlB4 powders b) Lattice parameters, and c) unit cell volume as a function of Cr substitution. Solid lines and symbols denote experimental LPs; dashed lines and open symbols, denote calculated LPs.
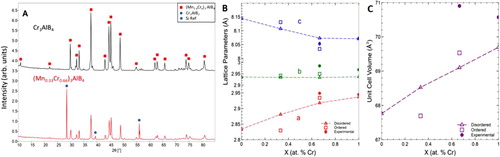
Table lists the LP and Vuc changes in the (Mn1-xCrx)2AlB2 compositions as a function of x. The same results are plotted alongside the calculated values for the disordered structure as well as Vegard’s law estimations in Figure (b) and (c). Though the calculated values are slightly smaller than experimental, the trends are identical for both sets of LPs. Here the largest deviation is found for the c-LP of Mn2AlB2 being −2.4%. The deviations are even les (−1.2% to −1.5%) for x ≠ 0. The deviations for a- and b-LPs are in the range 0 to −0.9% as compared to experiment. This agreement could likely be improved through a much more detailed investigation that includes more intricate magnetic configurations and, maybe, through the use of other xc-functionals.
From these results, it is clear, that the a-LP and Vuc deviate little from those expected from Vegard’s law. In contradistinction, the b- and c-LPs deviate from Vegard’s law, with a negative deviation in the b-direction and a positive deviation in the c-direction. Given that these deviations are theoretically predicted, it follows that these trends are embedded in the intricacies of bonding [Citation17]. Based on both the theoretical and experimental results, it is reasonable to assume that the solid solution range extends over all compositions. This is consistent with the fact that Mn2AlB2 and Cr2AlB2 both crystalize in the Cmmm space group.
(Mn1-xCrx)3AlB4
After reacting, the (Mn1-xCrx)3AlB4 composition did not densify, instead, the powders were loosely bound with a ‘burnt toast’ like consistency. XRD analysis of the powders showed that the mixture was fully reacted and indeed formed a 314 solid solution with 16.5 ± 2.6 wt. % of Cr2AlB2 impurities (Figure (a)). Compared to Cr3AlB4 synthesized in the same manner, there is evidence of peak shifting, indicating the successful formation of a solid solution [Citation30]. Table lists the LPs and the Vuc derived from these patterns. The experimental and calculated LP and Vuc values are shown for both the disordered and ordered (Mn1-xCrx)3AlB4 compositions in Figure (b) and (c), respectively Like in the (Mn1-xCrx)2AlB2 case, the calculated LPs are lower than the experimental, however trends from all three can be analysed. In the disordered model, as the Cr is substituted into the system, the b-LP remains mostly unchanged. Concomitantly there is expansion in the a-direction and contraction in the stacking, or c-direction. This trend parallels that of the 212 system, with regards to the contraction in the stacking direction and expansion perpendicular to the direction of the B chains.
Overall, the calculated values show a monatomic increase in Vuc as a function of Cr loading. The experimental results, however, show the opposite trend. At this time, the reasons for the discrepancy are not understood and more theoretical work is indicated. This comment notwithstanding the differences in the absolute values between theory and experiment are, at worst, <2%. As important, the fact that theory predicts the ordered phases to be unstable, suggests they are not ordered. More work, possibly neutron diffraction that can differentiate between Cr and Mn, is needed to resolve this important question.
On the other hand, attempts to synthesize the (Mn0.66Cr0.33)3AlB4 solid solutions resulted in a mixture of Mn2AlB2 and Cr2AlB2. Said otherwise, like theory predicts, (Mn0.66Cr0.33)3AlB4 is thermodynamically unstable at this stoichiometry at the processing temperature used, viz. 1050°C. Dai et al. [Citation17] predicted Mn2CrAlB4 to be stable. This prediction is, as noted above and the results shown here, is incorrect because the authors did not include the M2AlB2 phases in their analysis.
Conclusions
Herein we show, for the first time, that in the (Mn1-xCrx)2AlB2 system, a continuous M-site substitution is possible, for all x values. First-principles studies of disordered solid solution thermodynamics and experimental synthesis both confirm the stability of the system across the entire range. While the experimental values for the lattice parameter and unit cell volume are slightly larger than the calculated values, both exhibit similar trends. Similarly, the shifts in the a- and UC volume follow Vegard’s law, but b- and c-LPs, do not, indicating a negative and positive deviation, respectively. Investigations of the magnetic and electronic behaviour of such MAB solid solutions, with full range M-site substitution, is a topic of future work.
In the (Mn1-xCrx)3AlB4 system, the expected stability was modelled for both a disordered and ordered structure across the entire range. Experimentally, a solid solution of (Mn0.33Cr0.66)3AlB4 phase was successfully synthesized, again for the first time, while attempts to make (Mn0.66Cr0.33)3AlB4 were unsuccessful, paralleling the findings of our DFT calculations. These results indicate that future solid solutions may be possible for 314 MAB phases (M ≠ Mn) with a base of Cr3AlB4.
Acknowledgements
The calculations were carried out using supercomputer resources provided by the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Centre (NSC), the High Performance Computing Center North (HPC2N), and the PDC Center for High Performance Computing.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Jeitschko W. Die Kristallstruktur von MoAlB. Monatshefte für Chemie und verwandte Teile anderer Wissenschaften. 1966;97(5):1472–1476.
- Becher H, Krogmann K, Peisker E. Über das ternäre borid Mn2AlB2. Zeitschrift für anorganische und allgemeine Chemie. 1966;344(3–4):140–147.
- Stadelmaier H, Burgess R, Davis H. The FE-AL-B ternary system. Metall. 1966;20(3):225–226.
- Chaban N, Kuz’ma Y. Izvestiya Academii Nauk SSSR. Neorg Mater. 1973;9:1696.
- Tan X, Chai P, Thompson CM, et al. Magnetocaloric effect in AlFe2B2: toward magnetic refrigerants from earth-abundant elements. J Am Chem Soc. 2013 Jun;135(25):9553–9557.
- Kota S, Zapata-Solvas E, Ly A, et al. Synthesis and characterization of an alumina forming nanolaminated boride: MoAlB. Sci Rep. 2016 May;6:26475.
- Alameda LT, Moradifar P, Metzger ZP, et al. Topochemical deintercalation of Al from MoAlB: stepwise etching pathway, layered intergrowth structures, and two-dimensional MBene. J Am Chem Soc. 2018;140(28):8833–8840.
- Zhang H, Dai F-Z, Xiang H, et al. Phase pure and well crystalline Cr2AlB2: a key precursor for two-dimensional CrB. J Mater Sci Technol. 2019;35(8):1593–1600.
- Kota S, Sokol M, Barsoum MW. A progress report on the MAB phases: atomically laminated, ternary transition metal borides. Int Mater Rev. 2019;65(4):226–255.
- Rogl P. On the crystallographic relationship between monoborides (CrB, α-MoB) and actinide-borocarbides (UBC, ThBC). J Nucl Mater. 1979;80(1):187–189.
- Ade M, Hillebrecht H. Ternary borides Cr2AlB2, Cr3AlB4, and Cr4AlB6: the first members of the series (CrB2)nCrAl with n = 1, 2, 3 and a unifying concept for ternary borides as MAB-phases. Inorg Chem. 2015 Jul;54(13):6122–6135.
- Khazaei M, Wang J, Estili M, et al. Novel MAB phases and insights into their exfoliation into 2D MBenes. Nanoscale. 2019;11(23):11305–11314.
- Yu Y. Torsten Lundstrom crystal growth and structural investigation of the new quaternary compound Mo1-xCrxAlB with x = 0.39. J Alloys Compd. 1995;226:5–9.
- Okada S, Iizumi K, Kudaka K, et al. Single crystal growth of (MoXCr1-X)AlB and (MoXW1-X) AlB by metal Al solutions and properties of the crystals. J Solid State Chem. 1997;133:36–43.
- Du Q, Chen G, Yang W, et al. Magnetic frustration and magnetocaloric effect in AlFe2−xMnxB2(x = 0–0.5) ribbons. J Phys D: Appl Phys. 2015;48:335001.
- Hirt S, Yuan F, Mozharivskyj Y, et al. AlFe2-xCoxB2 (x = 0-0.30): TC tuning through co substitution for a promising magnetocaloric material realized by spark plasma sintering. Inorg Chem. 2016 Oct;55(19):9677–9684.
- Dai F-Z, Xiang H, Sun Y, et al. M2m′alb4 (M = Mn, Fe, Co, M′= Cr, Mo, W): theoretical predicted ordered MAB phases with Cr3AlB4 crystal structure. J Mater Sci Technol. 2019;35(7):1432–1438.
- Blöchl PE. Projector augmented-wave method. Phys Rev B. 1994;50(24):17953–17979.
- Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B. 1999;59(3):1758–1775.
- Kresse G, Hafner J. Ab initio molecular dynamics for liquid metals. Phys Rev B. 1993 Jan;47(1):558–561.
- Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput Mater Sci. 1996;6(1):15–50.
- Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Physical Review B. 1996;54(16):11169–11186.
- Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77(18):3865–3868.
- Monkhorst HJ, Pack JD. Special points for Brillouin-zone integrations. Phys Rev B. 1976;13(12):5188–5192.
- Zunger A, Wei SH, Ferreira LG, et al. Special quasirandom structures. Phys Rev Lett. 1990;65(3):353–356.
- Dahlqvist M, Alling B, Abrikosov IA, et al. Phase stability of Ti2AlC upon oxygen incorporation: a first-principles investigation. Phys Rev B. 2010;81(2):024111.
- Dahlqvist M, Alling B, Rosen J. Stability trends of MAX phases from first principles. Phys Rev B. 2010;81(22):220102.
- Thore A, Dahlqvist M, Alling B, et al. Temperature dependent phase stability of nanolaminated ternaries from first-principles calculations. Comput Mater Sci. 2014 August;91:251–257.
- Hubbard CR, Snyder RL. RIR-measurement and use in quantitative XRD. Powder Diffr. 1988;3(2):74–77.
- Kota S, Wang W, Lu J, et al. Magnetic properties of Cr2AlB2, Cr3AlB4, and CrB powders. J Alloys Compd. 2018;767:474–482.