
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Here, thermodynamic stability and lithium storage properties of double transition metal M2TiAlC2 o-MAX phases (M = Cr, V, Mo, Nb, Ta, Hf, Zr, Sc, Y, La) are theoretically investigated by density functional theory (DFT) calculation. M2TiAlC2 with a larger M atomic radius shows larger interlayer space that may benefit the Li-ion intercalation. A promising theoretical capacity of 276.87 mAh g-1 is predicted for Sc2TiAlC2. The low Li-ion diffusion barriers (0.57–0.64 eV) for M2TiAlC2 indicate the possibility to achieve fast Li-ion diffusion that is crucial for designing high-power batteries. This work provides opportunities to explore MAX phases as promising Li-ion storage materials.
GRAPHICAL ABSTRACT
IMPACT STATEMENT
This work investigates the thermostability and lithium storage properties of double transition metal o-MAX phases by DFT calculation and provides a guideline for exploring MAX phases for lithium storage applications.
Introduction
MAX phases are hexagonal nanolaminated ternary metal/ceramics with the general formula Mn+1AXn, where M is the transition metal, A is the main III to V group elements, X is C or N and typically n = 1, 2, 3, 4, etc. [Citation1,Citation2]. There are currently more than 150 MAX phases known, including solid solutions [Citation3,Citation4]. Due to the alternative stacking between the A layer and ceramic layer, MAX phases possess high conductivity and structural stability that is being considered for electrical contacts, sensors, protective coating, microelectromechanical systems, and high-temperature structural applications [Citation5–8]. In recent years, MAX phases are receiving increasing attention as precursor materials of two-dimensional MXenes that are extensively investigated as electrochemical energy storage materials [Citation9–13]. Although much less attention has been paid to MAX phases as lithium storage materials, several studies confirmed the promising potential of using MAX phases as lithium storage electrode materials. In 2016, Xu et al. [Citation14] reported on the enhanced lithium storage performance of Ti3SiC2 and Ti2SC MAX phases by reducing the particle size of the MAX phases, achieving a maximum capacity of 180 mAh g−1. Zhao et al. [Citation15] demonstrated Nb2SnC MAX phases with a lithium storage capacity of 234 mAh g−1 and proposed that the Sn atomic layer can contribute additional capacity through reversible Sn–Li alloying reaction. Recently, Wu et al. [Citation16] showed the Ti2SnC MAX phases are capable of delivering 735 mAh g−1 and Li et al. [Citation17] reported V2SnC MAX phases could obtain high gravimetric capacity up to 490 mAh g−1 and proposed a lithium storage mechanism with V2C-Li and Sn–Li reactions.
To achieve high specific energy for Li-ion batteries, developing novel anode materials with a high specific capacity, low delithiation potential, high cycling stability, and reversibility are highly demanded [Citation18–20]. MAX phases featured with layered structures and high electrical conductivities are believed to be promising lithium storage hosts. However, to the best of our knowledge, studies on MAX phases as an electrode for energy storage is scarce and the charge storage mechanism is not fully understood yet. Therefore, it is of great interest and significance to explore the lithium storage properties of MAX phases. DFT calculations are effective methods for theoretical investigation on materials’ properties. Zhu et al. [Citation21] predicted by DFT calculation that the stress-enhanced V2SC MAX phases could achieve good lithium storage properties and soft interlayer in MAX phases provide the possibility for lithium storage. However, other researchers found the interlayer space of MAX phases is too narrow to incorporate lithium and the lithiation formation energy is positive [Citation22,Citation23]. Therefore, more studies focused on the exfoliation or interlayer expansion to enhance the chances of lithium interaction [Citation24,Citation25].
Elemental doping and substitution, in many cases, are effective routes to tune the electronic properties of MAX phases [Citation26,Citation27]. Therefore, we speculate that the ordered M2TiAlC2 (M = Cr, V, Mo, Nb, Ta, Hf, Zr, Sc, Y, La) MAX phases with outer Ti atomic layers substitutional strategy may expand the interlayer space and achieve enhanced lithium storage properties. In the ceramic layer structure, the face-centered cubic (fcc) transition metal carbides are more stable. Thus, the Ti located inside of the ceramic layer can usually stabilize the structure [Citation28]. Combining the M element substitution and the stability of Ti carbide layer, it can be expected that the M2TiAlC2 may achieve high lithium storage properties and structural stability.
In this work, the structural stability and electronic properties of the M2TiAlC2 MAX phases (M = Cr, V, Mo, Nb, Ta, Hf, Zr, Sc, Y, La) with or without lithium intercalation are investigated by DFT calculations. The phonon density of states is employed to predict the lithium storage capacity by checking the stabilities of M2TiAlC2 containing the different numbers of Li-ions. The kinetic properties are evaluated for M2TiAlC2 by analyzing the migration pathways of Li-ion.
Results and discussion
For selecting MAX phases as promising electrode materials, the thermodynamic stability, lithiation formation energy, theoretical capacity, and Li-ion diffusion barrier are vital criteria and will be carefully screened step by step.
Stability of M2TiAlC2
The calculated lattice constants and formation energies of M2TiAlC2 are listed in , in which the values of Ti3AlC2 matched well with the previous works [Citation29]. With the outer M atomic size increasing, the a and c lattice constants increase from 2.904 to 3.460 Å and 17.793 to 22.704 Å, respectively. With the atom size differences of substitutional M and Ti increase, the formation energies of M2TiAlC2 correspondingly increase, leading to the enhancement of instability. However, according to the formation energies, substitutions of Zr, Hf, Sc, and Y not only have less influence on the thermodynamics but also expand their interlayer space effectively. The substitutions of Cr, V, Mo, Nb, and Ta result in the decrease of interlayer space, and these M2TiAlC2 MAX phases are not considered for further investigation due to the difficulty for lithium intercalation. Unstable (∼ −0.05 eV) La2TiAlC2 is not further discussed as well.
Table 1. Lattice constants, cell volume, and formation energies, , of M2TiAlC2 MAX phases (M = Cr, V, Mo, Ti, Nb, Ta, Hf, Zr, Sc, Y, and La).
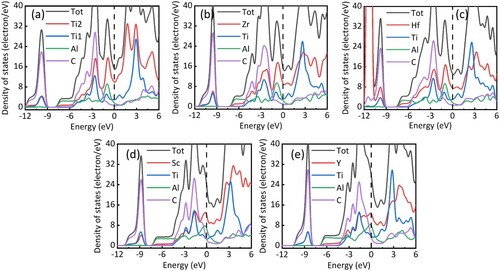
The density of states (DOS) of Ti3AlC2, Zr2TiAlC2, Hf2TiAlC2, Sc2TiAlC2, and Y2TiAlC2 phases are calculated as shown in . Similar to Ti3AlC2, the DOS of M2TiAlC2 has similar hybridization characteristics, including the covalent bonding of M and C and the metallic properties dominated by transition elements [Citation30]. Since the insufficient number of valence electrons for filling the bonding states, the hybridization peaks of Sc2TiAlC2 and Y2TiAlC2 will shift toward a higher energy level. The DOS in higher energy levels tend to fill nonbonding/antibonding states, which reduces the stability of the structure. It is in good agreement with the calculated formation energies. Since Ti, Zr, and Hf have the same number of valence electrons, the bonding states of M2TiAlC2 MAX phases are filled and their corresponding antibonding states remain unfilled, separating by a pseudogap in the Fermi level [Citation31]. Thus, the substitution of Sc and Y would be less stable than Zr, and Hf substitution. Besides, the order of phase stability is Ti3AlC2 > Zr2TiAlC2 > Hf2TiAlC2, which conforms to the order of M atomic size (Hf > Zr > Ti), indicating the M atom size is opposite to thermodynamic stability.
Figure 1. The density of states of (a) Ti3AlC2, (b) Zr2TiAlC2, (c) Hf2TiAlC2, (d) Sc2TiAlC2 and (e) Y2TiAlC2.
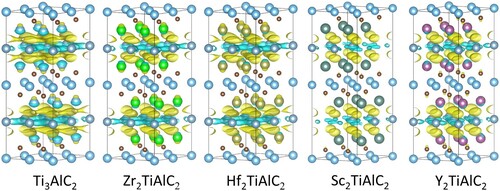
As shown in , the charge differences between Al and ceramic layers are calculated to obtain insight into the structural changes. The degree of the charge transfer represents bonding strength. Compared with Ti3AlC2, the M2TiAlC2 MAX phases show similar electronic distributions but less charge transfer, suggesting weaker M–Al bonding. The relatively weakening M–Al bonds represent the longer M–Al bonding length, further generating an expanding interlayer space [Citation32]. Besides, the weaker bonding strength can also be explained by overlapped states shifting toward higher energy levels as shown in . Combining the analysis of electronic structure and thermodynamics (formation energy), we reach the conclusion that the larger difference between the atomic sizes of M and Ti results in the larger interlayer space and the less structural stability, which agrees well with previous studies[Citation31,Citation33].
Figure 2. The charge differences of Ti3AlC2 and M2TiAlC2 MAX phases at the same isosurface with isovalue are 0.05 electrons/bohr3.
Lithium storage properties of M2TiAlC2
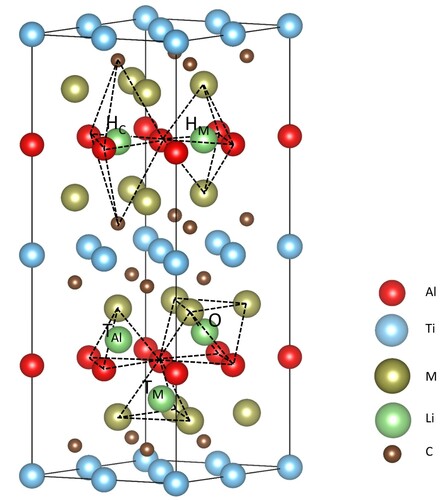
Characterized by the expanded interlayer space, M2TiAlC2 MAX phases are interesting to be explored as electrode materials for LIBs. To figure out the energetically favorable sites of Li-ion in M2TiAlC2, five configurations are considered as follows: (1) M3Al tetrahedron (TM), (2) Al3Ti tetrahedron (TAl), (3) M3Al3 octahedron (O), (4) C hexahedron (HC), (5) M hexahedron (HM), as shown in . The more negative value of formation energy is, the more energetically stable configuration is. The lithiation formation energies of TM, HM, and Hc are positive, indicating the Li-ion in these sites are unfavorable. Noticeably, Li in O sites are all moved to the surrounding HC after full optimizations, indicating the inserted Li should firstly be stored in the Al layer. Besides, the formation energy values of O and HC sites are considerably close, as listed in Table S1, which indicates HC is the most energetic favorable lithiation site.
Figure 3. Structure of M2TiAlC2 MAX phases and initial lithium storage sites, where the black dotted line represents different interstices.
To obtain insight into the lithiation process, the Bader charge and charge difference analyses are performed. In , the charge transfer of Li in M2TiAlC2 is around −0.72 |e| that loses more electrons than in Ti3AlC2 (−0.69 |e|), which indicates Li will be more ionic states in the large interlayer spaces. Meanwhile, Al gains more electrons that explain the increasing degree of Li–Al alloy reaction. The charge transfer of Ti remains almost unchanged, reflecting that lithiation has slight influences on the interior of the ceramic layer. Besides, charge differences show the interaction between Li and host material, as shown in Figure S1. The Coulomb force between Li and M elements is weaker with the interlayer space enlarging because the smaller positive electron cloud generates less repulsion.
Table 2. Charge transfer with reference to isolated atoms (in electrons; calculated by the Bader approach) for lithiation of M2TiAlC2 MAX phases.
Specific capacity is a crucial parameter that determines the energy density of LIBs. To confirm the theoretical specific capacities of M2TiAlC2, the lithiation formation energy for each continually inserting Li is calculated via Eq (S2) and presented in (a). Obviously, Ti3AlC2 shows the lowest
, indicating less thermodynamic stable for Li insertion. Once the value of
tends to be zero or positive, no more lithium can be inserted, which corresponds to the maximum saturation of Li storage. Base on the Eq (S3), the theoretical capacities of Ti3AlC2, Zr2TiAlC2, Hf2TiAlC2, Sc2TiAlC2, and Y2TiAlC2 are calculated to be 243.54, 252.55, 105.03, 411.22, and 256.7 mAh g−1, respectively. Furthermore, the dynamical stability of maximal Li insertions is investigated by performing lattice phonon calculations, as shown in Figure S2. Imaginary frequency demonstrates the structure is dynamically unstable and the partial phonon density of stats of ceramic layer and Al layer with Li-ions are calculated. Among all M2TiAlC2 MAX phases, the ceramic layer is calculated without imaginary frequency, suggesting the dynamically stable and the optical and acoustical branches are well-separated, presenting a phonon dispersion characteristic of layered material [Citation34]. Ti3AlC2 phases can maintain only one Li atom per Al layer in a supercell with an imaginary frequency value of 100 THz, which indicates the theoretical capacity would be lower than ∼122.84 mAh g−1 calculated via Eq (S3). However, the Sc2TiAlC2 could maintain two Li atoms per Al layer in a supercell without any imaginary frequency, and the theoretical capacity could be 276.87 mAh g−1 that is twice larger than Ti3AlC2. With the same Li storage number but different molar weights, the theoretical capacities of Zr2TiAlC2 and Hf2TiAlC2 are 169.39 and 105.03 mAh g−1, respectively. After lithiation, as shown in Figure S3, the conductivities of M2TiAlC2 have no significant change because the metallic property is contributed mainly by M elements’ d orbital.
Figure 4. (a) The lithiation formation energy for M2TiAlC2 for every inserting Li. The bonding length between the (b) M and Li, (c) Al and Al, (d) Al and Li as the function with the inserted Li number.
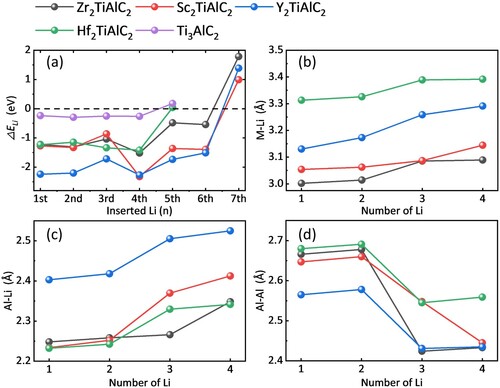
To clarify the effect of interlayer space on lithium storage, the M–Li, Al–Li, and Al–Al bonding lengths (representing different directions of interlayer space) are calculated, as shown in (b–d). The sudden change of the Al–Li and Al–Al bonding lengths happens at the third Li insertion. On the one hand, the M–Li bonding length changes slightly, reflecting that lithiation hardly influences the ceramic layer. On the other hand, the Al–Li boding length increases, and Al–Al bonding length decreases as the inserted Li number increases, indicating the Al layer atoms will rearrange during lithiation/delithiation. Al can rearrange in M2TiAlC2 easier because of the larger interlayer space that produces less Coulomb force and weaker M–Al bonding.
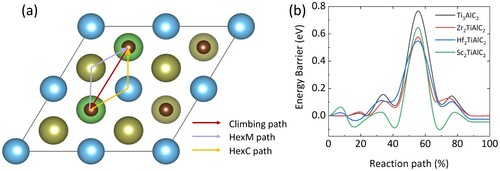
The charge/discharge rate capability of rechargeable LIBs is mainly determined by the ion diffusion kinetic. The intrinsic metallic behavior of M2TiAlC2 has been demonstrated by DOS [Citation35]. The NEB method is performed to estimate the Li diffusion. Three diffusion pathways along the high symmetry line between the two neighboring favorable sites (HC) are designed to find the minimum energy barrier, as shown in (a). However, the diffusion barrier of the HexM path is around 1.2 eV, which implicates M repulsive force hinders this diffusion path. Besides, the climbing path could not be realized because the ceramic layer’s repulsive force is larger than the Al layer. The diffusion energy barriers of the HexC path are relatively low, as presented in (b). The minimum energy barrier is 0.57 eV in Hf2TiAlC2. It is worth noting that along the HexC path, the diffusion trajectory of Li-ion passes through an Al position, resulting in the position derivation of the Al atom. Hence, if there is enough space for Al to move, the Li-ion would diffuse more easily. As demonstrated above, M substitution expands the interlayer space that facilitates the easy rearrangement of Al, thereby reducing the Li-ion diffusion barriers in M2TiAlC2.
Figure 5. (a) The three types of Li diffusion pathways and (b) the diffusion energy barrier for M2TiAlC2.
Conclusions
We performed a computational investigation on the structural and lithium storage properties of M2TiAlC2 MAX phases. DFT calculations demonstrate that M2TiAlC2 MAX phases are thermally stable. M elements substituting expands the interlayer space expands and thus reduces lithiation formation energy, and the charge difference shows that the strong repulsion force decreases, suggesting the great potential for lithium storage. Next, the maximum theoretical capacities of the M2TiAlC2 MAX phases are calculated. Zr2TiAlC2 and Sc2TiAlC2 are predicted with the maximum specific capacities of 169.39 and 276.87 mAh g−1 respectively. Finally, the different diffusion pathways were screened for investigating the diffusion barriers of Li-ion into M2TiAlC2 MAX phases. The lowest diffusion energy barrier (∼0.57 eV) is obtained for Hf2TiAlC2.
In this work, the M elements substitution strategy was proposed for fine-tuning the interlayer space and electronic structure of MAX phases as promising lithium storage materials. The guideline to improve the lithium storage properties of MAX phases are summarized as following: (1) enlarge the interlayer space by larger radii elements substitution; (2) select proper A elements to make inserted Li in more ionic states.
Supplemental Material
Download MS Word (10.1 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Barsoum MW. MAX phases: properties of machinable ternary carbides and nitrides. Weinheim: Wiley-VCH; 2013.
- Barsoum M. The MN+1AXN phases. A new class of solids; thermodynamically stable nanolaminates. Prog Solid State Chem. 2000;28:28–201.
- Naguib M, Bentzel GW, Shah J, et al. New solid solution MAX phases: (Ti0.5, V0.5)3AlC2, (Nb0.5, V0.5)2AlC, (Nb0.5, V0.5)4AlC3 and (Nb0.8, Zr0.2)2AlC. Mater Res Lett. 2014;2(4):233–240.
- Sokol M, Natu V, Kota S, et al. On the chemical diversity of the MAX phases. Trends in Chemistry. 2019;1(2):210–223.
- Xu J, Qiu N, Huang Q, et al. Theoretical investigations on structural and thermo-mechanical properties of layered ternary carbide Th–Al–C systems. J Nucl Mater. 2020;540:152358.
- Hajas DE, to Baben M, Hallstedt B, et al. Oxidation of Cr2AlC coatings in the temperature range of 1230 to 1410°C. Surf Coat Technol. 2011;206(4):591–598.
- Tallman DJ, Hoffman EN, EaN C, et al. Effect of neutron irradiation on select MAX phases. Acta Mater. 2015;85:132–143.
- Lane NJ, Vogel SC, EaN C, et al. High-temperature neutron diffraction and first-principles study of temperature-dependent crystal structures and atomic vibrations in Ti3AlC2, Ti2AlC, and Ti5Al2C3. J Appl Phys. 2013;113(18):183519.
- Li Y, Shao H, Lin Z, et al. Author correction: a general Lewis acidic etching route for preparing MXenes with enhanced electrochemical performance in non-aqueous electrolyte. Nat Mater. 2021;20(4):571–571.
- Zhang X, Zhang Z, Zhou Z. MXene-based materials for electrochemical energy storage. J Energy Chem. 2018 Jan;27(1):73–85.
- Tang Q, Zhou Z, Shen P. Are MXenes promising anode materials for Li ion batteries? Computational studies on electronic properties and Li storage capability of Ti3C2 and Ti3C2X2 (X = F, OH) monolayer. J Am Chem Soc. 2012;134(40):16909–16916.
- Miao N, Wang J, Gong Y, et al. Computational prediction of boron-based MAX phases and MXene derivatives. Chem Mater. 2020;32(16):6947–6957.
- Barmann P, Haneke L, Wrogemann JM, et al. Scalable synthesis of MAX phase precursors toward titanium-based MXenes for lithium-ion batteries. ACS Appl Mater Interfaces. 2021;13(22):26074–26083.
- Xu J, Zhao M-Q, Wang Y, et al. Demonstration of Li-ion capacity of MAX phases. ACS Energy Lett. 2016;1(6):1094–1099.
- Zhao S, Dall’Agnese Y, Chu X, et al. Electrochemical interaction of Sn-containing MAX phase (Nb2SnC) with Li-ions. ACS Energy Letters. 2019;4(10):2452–2457.
- Wu H, Zhu J, Liu L, et al. Intercalation and delamination of Ti2SnC with high lithium ion storage capacity. Nanoscale. 2021;13(15):7355–7361.
- Li Y, Ma G, Shao H, et al. Electrochemical lithium storage performance of molten salt derived V2SnC MAX phase. Nanomicro Lett. 2021;13(1):158.
- Sun L, Xie J, Zhang L, et al. 2D black TiO2-x nanoplate-decorated Ti3C2 MXene hybrids for ultrafast and elevated stable lithium storage. FlatChem; 2020;20:100152.
- Zhu G, Chen T, Wang L, et al. High energy density hybrid lithium-ion capacitor enabled by Co3ZnC@N-doped carbon nanopolyhedra anode and microporous carbon cathode. Energy Storage Mater. 2018;14:246–252.
- Chen T, Cheng B, Chen R, et al. Hierarchical ternary carbide nanoparticle/carbon nanotube-inserted N-doped carbon concave-polyhedrons for efficient lithium and sodium storage. ACS Appl Mater Interfaces. 2016;8(40):26834–26841.
- Zhu J, Chroneos A, Wang L, et al. Stress-enhanced lithiation in MAX compounds for battery applications. Appl Mater Today. 2017;9:192–195.
- Filippatos PP, Hadi MA, Christopoulos SRG, et al. 312 MAX phases: elastic properties and lithiation. Materials (Basel). 2019;12(24):4098.
- Sun W, Shah SA, Chen Y, et al. Electrochemical etching of Ti2AlC to Ti2CTx (MXene) in low-concentration hydrochloric acid solution. J Mater Chem A. 2017;5(41):21663–21668.
- Tesfaye AT, Mashtalir O, Naguib M, et al. Anodized Ti3SiC2 As an anode material for Li-ion microbatteries. ACS Appl Mater Inter. 2016;8(26):16670–16676.
- Chen X, Zhu Y, Zhu X, et al. Partially etched Ti3AlC2 as a promising high-capacity lithium-ion battery anode. ChemSusChem. 2018;11(16):2677–2680.
- Khazaei M, Wang V, Sevik C, et al. Electronic structures of iMAX phases and their two-dimensional derivatives: a family of piezoelectric materials. Phys Rev Mater. 2018;2(7):074002.
- Cover MF, Warschkow O, Bilek MMM, et al. A comprehensive survey of M2AX phase elastic properties. J Phys: Condens Matter. 2009;21(30):305403.
- Anasori B, Xie Y, Beidaghi M, et al. Two-dimensional, ordered, double transition metals carbides (MXenes). ACS Nano. 2015;9(10):9507–9516.
- Magnuson M, Mattesini M. Chemical bonding and electronic-structure in MAX phases as viewed by X-ray spectroscopy and density functional theory. Thin Solid Films. 2017;621:108–130.
- Sun Z, Music D, Ahuja R, et al. Bonding and classification of nanolayered ternary carbides. Phys Rev B. 2004;70(9):092102.
- Bouhemadou A, Khenata R. Structural, electronic and elastic properties of M2SC (M = Ti, Zr, Hf) compounds. Phys Lett A. 2008;372(42):6448–6452.
- Brown ID, Shannon RD. Empirical bond-strength–bond-length curves for oxides. Acta Crystallogr A. 1973;29(3):266–282.
- Khazaei M, Arai M, Sasaki T, et al. Trends in electronic structures and structural properties of MAX phases: a first-principles study on M2AlC (M = Sc, Ti, Cr, Zr, Nb, Mo, Hf, or Ta), M2AlN, and hypothetical M2AlB phases. J Phys: Condens Matter. 2014;26(50):505503.
- Zhang X, Yu Z, Wang S-S, et al. Theoretical prediction of MoN2 monolayer as a high capacity electrode material for metal ion batteries. J Mater Chem A. 2016;4(39):15224–15231.
- Wang D, Gao Y, Liu Y, et al. First-principles calculations of Ti2N and Ti2NT2 (T = O, F, OH) monolayers as potential anode materials for lithium-ion batteries and beyond. J Phys Chem C. 2017;121(24):13025–13034.