
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
We identify the state-associated cohesive energy and band filling as descriptors to quantify the site-to-site chemical bonding and magnetic moments of high-entropy alloys (HEAs). We find that the s-state cohesive energy is indispensable in determining the bonding-strength trend while the s-band filling is effective in determining the magnetic moments of CrMnFeCoNi HEAs. This unusual behavior stems from the localized and transferred itinerant electrons. Our study establishes a fundamental physical picture of chemical-bonding and magnetic interactions of HEAs and helps design advanced structural alloys.
GRAPHICAL ABSTRACT
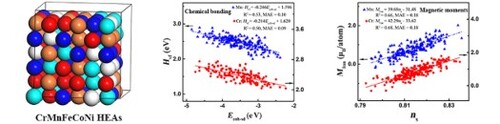
Introduction
High-entropy alloys (HEAs) often exhibit highly desirable combinations of mechanical and functional properties that are promising for many practical applications [Citation1–6], because of their disordered multicomponent structures. CrMnFeCoNi HEAs, one of the most promising HEAs to date, possess the unique site-to-site chemical and magnetic disorder and exhibit superior damage tolerance and fracture toughness [Citation7–11]. However, the nearly randomly distributed atomic species of HEAs limit understanding the origin of their unique mechanical behavior. Therefore, a fundamental issue for HEAs in general and for CrMnFeCoNi in particular is how to estimate quantitatively the local chemical-environment and magnetic effects.
Several important attempts have been made to address this issue. Atomic-scale short-range order (SRO) parameters are widely used in studying the chemical and magnetic distribution of HEAs [Citation12]. However, they are mainly suitable for describing the statistical properties instead of the site-to-site chemical bonding and magnetic moments. The magnetic frustration of anti-ferromagnetic Cr atoms is expected to reduce the Cr–Cr pairs in the nearest-neighbor (1nn) shell of CrCoNi and CrMnFeCoNi alloys [Citation13–18], while the strong chemical bonding of Cr–Cr pairs should promote the structural stability. There thus seems to exist a competition between the magnetic frustration and chemical bonding of the Cr–Cr pairs in determining the structure of CrMnFeCoNi. Furthermore, the widely used Slater-Pauling curve is mainly applicable to characterize the average magnetic moments of transition metals (TMs) and their bi-metallics [Citation19–21] but not those of HEAs [Citation22]. In addition, the valence-electron concentration, electronegativity difference, atomic-size difference, and mixing entropy (enthalpy) are also proposed as descriptors to study the average bulk-related properties of HEAs like phase stability and deformation mechanism [Citation2,Citation23–26] but are inconvenient to capture the local chemical-environment and magnetic effects.
Herein we propose the mixed d- and s-state cohesive energy and the s-band filling based on the tight-binding (TB) and Friedel models as novel descriptors for characterizing the local chemical bonding and magnetic moments of CrMnFeCoNi HEAs. We find that the s-state properties are effective and indispensable in determining the bonding characteristics and local magnetic moments of CrMnFeCoNi HEAs, which stem from the localized and transferred itinerant electrons.
Materials and methods
The DFT calculations were performed via Vienna Ab initio Simulation Package (VASP) [Citation27] with the projector augmented wavefunctions (PAW) potentials [Citation28] and Perdew−Burke−Ernzerhof (PBE) functional [Citation29]. Both the non-spin-polarized and spin-polarized calculations were carried out to study the chemical environments and magnetic moments of HEAs. More computational details are shown in Note S1.
Results and discussion
The chemical bonding and magnetic moments of HEAs likely stem from the coupling of atoms’ different states. For TMs and their bi-metallics, the TB and Friedel models [Citation30,Citation31] demonstrate that the chemical bonding is mainly controlled by the d-band width (the cohesive energy), while the Slater-Pauling curve [Citation19,Citation20] indicates that the average magnetic properties are governed by the d-band filling. We thus employ the state-associated cohesive properties and band filling to characterize the chemical bonding and magnetic effects of HEAs, which are the cohesive energy Ecoh-t and band filling nt and their s- and d-state components (Ecoh-s, Ecoh-d, ns, and nd) (Note S2 and Table S1). For a given site in HEAs, its complex chemical-environment and magnetic effects are quantified with the geometric mean of Ecoh-t and nt of its surrounding atoms, as,
(1)
(1)
(2)
(2) where
and
are the cohesive energy and band filling of ith involved atom in the bulk state and N is the number of the involved atoms. Similarly, the corresponding s- and d-state contribution as well as the mixed s- and d-state contribution can be also obtained. Accordingly, one can define the descriptors for the 1nn shell and other-ranged shells of a given site in HEAs.
To study the chemical-environment effect of HEAs, we focus on the non-spin-polarized vacancy formation energy (Hvf) of CrMnFeCoNi to exclude the contribution of magnetic properties. Figure plots the correlation between Hvf and the cohesive-energy descriptors (obtained with atoms in the vacancies’ 1nn shell). Apparently, Hvf shows a linear relation with the d-state cohesive energy Ecoh-d (but not the s-state cohesive energy Ecoh-s) for Fe, Co, and Ni vacancies with reasonable accuracy (Figure a and b and Fig. S1a and b). Thus, the d-states dominate the chemical-environment effect of Fe, Co, and Ni sites in CrMnFeCoNi, compatible with the TB and Friedel models [Citation30,Citation31]. However, both Ecoh-d and Ecoh-s perform poorly in describing the Hvf of Cr and Mn vacancies (Figure c and Fig. S1c). Interestingly, Ecoh-sd (involving the s-state cohesive energy of 1nn Cr atoms and the d-state cohesive energy of other 1nn atoms) describes well the Hvf of Cr and Mn sites (Figure d). Note that we also attempt to use the s-state cohesive energy of other components such as Mn to describe the Hvf since Mn is also anti-ferromagnetic in the ground state like Cr, however, the mixed cohesive-energy descriptor involving the Mn atoms’ s-states and other atoms’ d-states fails to capture the Hvf trends of Cr and Mn sites (Fig. S1d). This seems to contradict the conventional TB and Friedel models. For the interatomic bonding, the Cr atoms’ behavior depends strongly on their surrounding atoms in CrMnFeCoNi: the Cr atoms bond to Fe, Co, and Ni atoms mainly with the d-states and likely bond to Cr and Mn atoms mainly with the s-states. The underlying mechanism will be discussed in detail below.
Figure 1 The non-spin-polarized Hvf against the state-associated cohesive energies in CrMnFeCoNi HEAs. a and b, The Fe, Co and Ni’s Hvf against the Ecoh-d. c and d, Comparison between Ecoh-d and Ecoh-sd in describing Cr and Mn’s Hvf. e, The Fe, Co, Cr and Mn’s Hvf against the combined descriptor DCE.
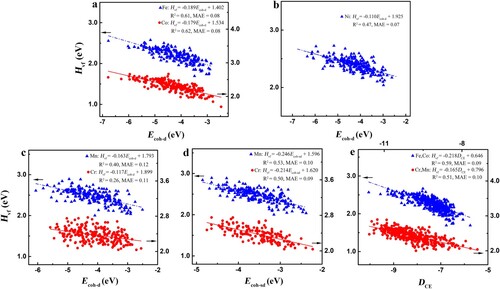
Furthermore, the Hvf of different vacant species can be correlated by combining the removed vacant atoms’ cohesive energies (Ecoh, rva) and their environment descriptors (Ecoh, 1nn), as DCE = k1 × Ecoh, rva + k2 ×Ecoh, 1nn, where k1 and k2 reflect the coupling between the removed vacant atom and its nearest neighbors. Figure e shows the relation of Hvf against DCE: except Ni sites, Fe and Co sites’ Hvf comply with a consistently linear relation, whereas Cr and Mn sites do another one. This implies that Fe atoms are similar to Co atoms in the bonding characteristics of CrMnFeCoNi. In addition, the bonding-strength trends of Cr and Mn atoms are also similar although the d-band properties of Cr atoms change in CrMnFeCoNi compared with their pure bulk states while those of Mn atoms remain unchanged.
These findings clarify several crucial controversies and have important implications for the chemical bonding of HEAs. Figure shows that Hvf of CrMnFeCoNi linearly decreases with increasing of the cohesive-energy descriptor, implying that the sites with more small-cohesive-energy (the large absolute value) nearest neighbors should have enhanced stability. In particular, the component of the cohesive-energy descriptors is in the order of Ecoh-d, Mn < Ecoh-d, Fe < Ecoh-s, Cr < Ecoh-d, Co < Ecoh-d, Ni for the Cr sites, different from that of the bulk TM d-state cohesive energy. Note that the Cr–Cr pairs are found to present magnetically repulsive interactions [Citation13–18]. Hence, Cr–Cr pairs are detrimental to the stability of CrMnFeCoNi from the view of both chemical bonding and magnetic frustration, eliminating the generally considered contradiction that chemical bonding and magnetic frustration of Cr–Cr pairs compete with each other. On the other hand, the sites with more large-cohesive-energy (the small absolute value) nearest neighbors like Ni are more prone to form vacancies in CrMnFeCoNi, in agreement with the findings that vacancies in CrCoNi, CrCoFeNi, and CrMnFeCoNi alloys are more likely to generate at the sites with a Ni-rich 1nn shell [Citation32,Citation33]. Overall, the nearest neighbors of a given site of HEAs significantly affect its stability, supporting the existence of SRO. Notably, the SRO parameters are generally adopted to understand the statistical average properties of HEAs [Citation12–18], whereas our scheme effectively quantifies the local chemical bonding of HEAs by considering the bonding strength between a given atom and its nearest neighbors.
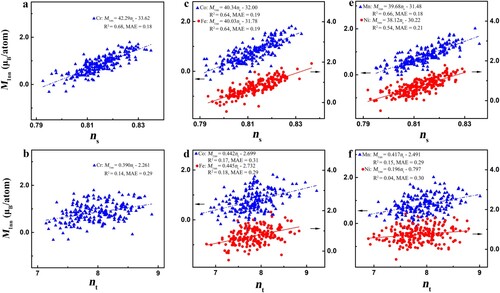
We now turn to understand the magnetism of CrMnFeCoNi HEAs. To elucidate the local magnetic effect, a 108-atom supercell is divided into 27 different tetrahedrons, which are the minimum repeat unit of an fcc crystal (Fig. S2). Figure and Table S2 show the correlation between the average magnetic moments of each tetrahedron (Mtetra) and the corresponding band-filling descriptors. Interestingly, the s-band filling ns instead of the total band filling nt is effective in determining the local magnetic moments of CrMnFeCoNi. Furthermore, we also study the average magnetic moments of the atoms around a vacancy site in CrMnFeCoNi. Remarkably, the local magnetic moments around vacancies (vacancies’ 1nn atoms, M1nn) are still well-characterized by ns rather than nt (Figure ). In addition, the average magnetic moments of atoms in the vacancies’ second-nearest-neighbor shell (M2nn), third-nearest-neighbor shell (M3nn), fourth-nearest-neighbor shell (M4nn), and long-range shell (Mlong) are also linearly related to the corresponding ns (Fig. S3). This is particularly encouraging, as the complex magnetic ordering of CrMnFeCoNi can be quantitatively captured by a simple electronic descriptor. Moreover, by linearly combining the environment-effect descriptor Ecoh,1nn and magnetic moments, we demonstrate that the magnetic moments exhibit the different range-reaching properties in determining the spin-polarized Hvf, depending on the surrounding chemical distributions of vacancies (Note S3, Fig. S4 and Table S3). Namely, the chemical distributions control the role of magnetic effects in the stability of CrMnFeCoNi, and thus both the chemical bonding and magnetic effects determine the SRO of HEAs.
Figure 2 Comparison between ns and nt in describing the local magnetic moments of CrMnFeCoNi HEAs. a-f, Tetrahedrons of six different bulk structures T1–T6.
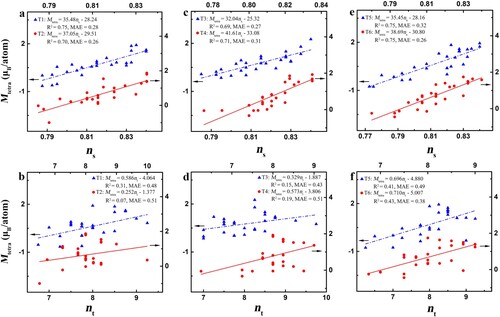
Figure 3 Comparison between ns and nt in describing the average magnetic moments of each vacancy’s 1nn-shell atoms of CrMnFeCoNi HEAs. a and b, Cr vacancies. c and d, Co and Fe vacancies. e and f, Mn and Ni vacancies.
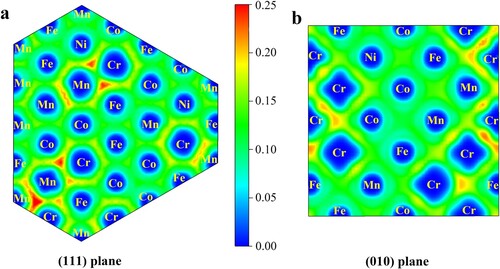
Our findings uncover a novel physical picture of the chemical-environment and magnetic nature of HEAs, which can be understood in the framework of the itinerant electron model [Citation34]. The itinerant electrons, different from the localized d-electrons and free-electron-like s-electrons, refer to the d-electrons located around the Fermi level and are widely used to explain the magnetism of TMs and their bi-metallics. The electronic localization function (ELF) [Citation35] shows that there are electrons localized mainly between Cr and Cr atoms and between Cr and Mn atoms but not between other atoms in CrMnFeCoNi (Figure ). In particular, there is no obvious electron localization between Mn atoms and non-Cr atoms such as Fe, Co, Ni and Mn atoms. Accordingly, these localized electrons generate a covalent-like bonding for Cr–Cr pairs and Cr-Mn pairs, but not for the other atom pairs such as Cr-Fe, Cr-Co, Cr-Ni, Mn-Mn, Mn-Fe, Mn-Co, and Mn-Ni pairs. Consequently, the mixed metallic and covalent-like bonding for Cr–Cr pairs and Cr-Mn pairs makes the d-state cohesive energy of Cr atoms in Cr–Cr pairs and Cr-Mn pairs increase from ∼ −8 eV to ∼ −4 eV, which is just similar to the value of s-state cohesive energy Ecoh-s of bulk Cr atoms. Meanwhile, the other atom pairs experience a less change and their component atoms exhibit the d-state bonding highly similar to that in the bulk state. This explains well the proposed cohesive-energy descriptors for the chemical-environment effect of different sites in CrMnFeCoNi (Figure ). Overall, it is the localized electrons that change the d-states of Cr–Cr pairs and Cr-Mn pairs and the d-states of atoms in CrMnFeCoNi still determine the chemical-environment effect, complying with the TB and Friedel models [Citation30,Citation31].
Figure 4 The electron localization function ELF of CrMnFeCoNi HEAs projected on (111) (a) and (010) (b) planes. The region with the deeper red color indicates the more electrons accumulated.
The Slater-Pauling curve demonstrates that the average magnetic moments of TMs and bi-metallics are determined by the d-band filling [Citation21,Citation22], which seems to contradict our results in CrMnFeCoNi HEAs. This can also be elucidated by the localized electrons between Cr and Cr atoms and between Cr and Mn atoms, which most likely stem from a part of itinerant electrons. The electron localization of Cr and Mn decreases the number of itinerant electrons and induces the energy level splitting and pronounced magnetic moments of Cr and Mn. In contrast, Fe, Co and Ni experience a less change of itinerant electrons as well as a minor change of magnetic moments. The Bader charge analysis [Citation36] shows that Co, Ni, and Fe atoms accept itinerant electrons from Cr and Mn atoms, and their electron-transfer amount reflects the change of their average magnetic moments (Table S4). In addition, by analyzing the local magnetic moments of atoms in CrMnFeCoNi, we find that Cr atoms always prefer an anti-ferromagnetic order while Mn atoms tend to have both the ferromagnetic and anti-ferromagnetic states in the lowest-energy configurations, consistent with the findings by George et al [Citation14] and Wang et al [Citation37]. This indicates that Cr and Mn atoms play different roles in determining the local magnetic properties of CrMnFeCoNi HEAs: Cr atoms favor maintaining their own anti-ferromagnetic order despite the variant chemical environments whereas Mn atoms’ magnetic moments are prone to be affected by the surrounding environments. Indeed, it has been demonstrated that the chemical environments present different effects on the magnetic moments of Cr and Mn atoms. When adding Al atoms into CrMnFeCo and MnFeCoNi alloys, the anti-ferromagnetic Mn atoms are suppressed to favor a ferromagnetic order but the magnetic order of anti-ferromagnetic Cr atoms remains almost unchanged [Citation37,Citation38]. The density of states (DOS) also shows that Cr and Mn atoms donate electrons while Co and Ni atoms mainly accept electrons compared to their pure bulk states (Fig. S5). It is noteworthy that for Cr and Mn atoms the up-spin electrons of anti-ferromagnetic atoms and the down-spin electrons of ferromagnetic atoms mainly participate in the itinerant-electron transfer. Accordingly, the up-spin electrons of Cr atoms contribute crucially to the transfer of itinerant electrons, whereas either the up- or down-spin electrons of Mn atoms do the case, depending on their specific magnetic order. This likely corresponds to the different magnetic behavior of Cr and Mn atoms. Indeed, the local magnetic-moment trends can be approximately captured by the number of Cr atoms (but not that of Mn atoms) for CrMnFeCoNi HEAs (Fig. S6) and this dominant effect is not due to the significant change of Cr atoms’ magnetic moments. Ritchie et al [Citation18] proposed that the magnetic moments of CrCoNi alloys are mainly related to the SRO of Cr atoms. Hence, Cr atoms govern the magnetic moments of both CrCoNi and CrMnFeCoNi alloys. Overall, the localized and transferred itinerant electrons between different atoms control the magnetic properties of CrMnFeCoNi HEAs.
Furthermore, our scheme predicts that the atoms in alloys (without a strong magnetic frustration) are prone to bond with the strong-cohesive-energy elements, leading to the SRO of atom pairs with the same elements. This prediction indeed has been fulfilled by the SRO of Co–Co, Fe-Fe, Ni-Ni, and Cu-Cu pairs in AlCoFeNiCu and AlCoFeNi alloys [Citation39]. Moreover, our scheme also suggests that the atoms with similar bonding characteristics are more likely to bond with each other such as the s- and s-state bonding and the d- and d-state bonding. Hence, Al prefers to bond with Cu but not Co, Fe, and Ni in AlCoFeNiCu HEAs as the s-states dominate the bonding of Al and Cu atoms (Al doesn’t have d-states and the d-states of Cu are fully filled) [Citation39]. On the other hand, both the magnetic frustration and cohesive bonding suppress the Cr–Cr pairs, making Cr atoms bond with the Co and Ni atoms in CrCoNi alloys. This rationalizes the SRO of Cr-Co and Cr-Ni pairs instead of Cr–Cr, Co–Co and Ni-Ni pairs in CrCoNi alloys [Citation12,Citation16]. Overall, our scheme can be alternative to the SRO parameters and the alloy-design strategies based on the SRO comply with the predictions of our scheme.
Conclusions
In summary, we have identified the mixed d- and s-state cohesive energy and s-band filling as effective descriptors in determining the chemical bonding and magnetic moments of CrMnFeCoNi HEAs, based on the TB and Friedel models. The effectiveness of s-state properties is due to the localized and transferred itinerant electrons in CrMnFeCoNi HEAs. These results build a novel picture for the chemical bonding and magnetic moments of HEAs, which resolves the controversy between the chemical bonding and magnetic frustration of Cr–Cr pairs in CrMnFeCoNi HEAs, outlines the principles governing the elusive local magnetic moments of HEAs, and supports the existence of SRO. Our descriptors can be alternative to the widely used SRO parameters by estimating the site-to-site bonding and magnetic moments and serve as a design tool to engineering HEAs’ properties.
Impact statement
The proposed state-associated cohesive energy and band filling are effective in determining the site-to-site chemical bonding and magnetic moments of CrMnFeCoNi, which can be alternative to the short-range order parameters.
Supplemental Material
Download PDF (1.6 MB)Acknowledgements
This work is supported by the National Natural Science Foundation of China (Nos. 22173034, 11974128, 52130101), the Program for JLU (Jilin University) Science and Technology Innovative Research Team (No. 2017TD-09), and the computing resources of the High Performance Computing Center of Jilin University, China.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Yeh J-W, Chen S-K, Lin S-J, et al. Nanostructured high-entropy alloys with multiple principal elements: novel alloy design concepts and outcomes. Adv Eng Mater. 2004;6:299–303. doi:10.1002/adem.200300567
- Zhang Y, Zuo TT, Tang Z, et al. Microstructures and properties of high-entropy alloys. Prog Mater Sci. 2014;61:1–93. doi:10.1016/j.pmatsci.2013.10.001
- Ye YF, Wang Q, Lu J, et al. High-entropy alloy: challenges and prospects. Mater Today. 2016;19:349–362. doi:10.1016/j.mattod.2015.11.026
- Miracle DB, Senkov ON. A critical review of high entropy alloys and related concepts. Acta Mater. 2017;122:448–511. doi:10.1016/j.actamat.2016.08.081
- George EP, Raabe D, Ritchie RO. High-entropy alloys. Nat Rev Mater. 2019;4:515–534. doi:10.1038/s41578-019-0121-4
- Zhang Y, Osetsky YN, Weber WJ. Tunable chemical disorder in concentrated alloys: defect physics and radiation performance. Chem Rev. 2022;122:789–829. doi:10.1021/acs.chemrev.1c00387
- Otto F, Dlouhý A, Somsen C, et al. The influences of temperature and microstructure on the tensile properties of a CoCrFeMnNi high-entropy alloy. Acta Mater. 2013;61:5743–5755. doi:10.1016/j.actamat.2013.06.018
- Gludovatz B, Hohenwarter A, Catoor D, et al. A fracture-resistant high-entropy alloy for cryogenic applications. Science. 2014;345:1153–1158. doi:10.1126/science.1254581
- Ma D, Grabowski B, Körmann F, et al. Ab initio thermodynamics of the CoCrFeMnNi high entropy alloy: importance of entropy contributions beyond the configurational one. Acta Mater. 2015;100:90–97. doi:10.1016/j.actamat.2015.08.050
- Troparevsky MC, Morris JR, Daene M, et al. Beyond atomic sizes and Hume-Rothery rules: understanding and predicting high-entropy alloys. JOM. 2015;67:2350–2363. doi:10.1007/s11837-015-1594-2
- Wang S, Zhang T, Hou H, et al. The magnetic, electronic, and thermodynamic properties of high entropy alloy CrMnFeCoNi: a first-principles study. Phys status solidi. 2018;255:1800306. doi:10.1002/pssb.201800306
- Tamm A, Aabloo A, Klintenberg M, et al. Atomic-scale properties of Ni-based FCC ternary, and quaternary alloys. Acta Mater. 2015;99:307–312. doi:10.1016/j.actamat.2015.08.015
- Niu C, Zaddach AJ, Oni AA, et al. Spin-driven ordering of Cr in the equiatomic high entropy alloy NiFeCrCo. Appl Phys Lett. 2015;106:161906. doi:10.1063/1.4918996
- Schneeweiss O, Friák M, Dudová M, et al. Magnetic properties of the CrMnFeCoNi high-entropy alloy. Phys Rev B. 2017;96:014437. doi:10.1103/PhysRevB.96.014437
- Zhang FX, Zhao S, Jin K, et al. Local structure and short-range order in a NiCoCr solid solution alloy. Phys Rev Lett. 2017;118:205501. doi:10.1103/PhysRevLett.118.205501
- Ding J, Yu Q, Asta M, et al. Tunable stacking fault energies by tailoring local chemical order in CrCoNi medium-entropy alloys. Proc Natl Acad Sci USA. 2018;115:8919–8924. doi:10.1073/pnas.1808660115
- Pei Z, Li R, Gao MC, et al. Statistics of the NiCoCr medium-entropy alloy: novel aspects of an old puzzle. npj Comput Mater. 2020;6:122. doi:10.1038/s41524-020-00389-1
- Walsh F, Asta M, Ritchie RO. Magnetically driven short-range order can explain anomalous measurements in CrCoNi. Proc Natl Acad Sci USA. 2021;118:e2020540118. doi:10.1073/pnas.2020540118
- Slater JC. The ferromagnetism of Nickel. II. temperature effects. Phys Rev. 1936;49:931–937. doi:10.1103/PhysRev.49.931.
- Pauling L. The Nature of the interatomic forces in metals. Phys Rev. 1938;54:899–904. doi:10.1103/PhysRev.54.899
- Lowitzer S, Ködderitzsch D, Ebert H, et al. Electronic transport in ferromagnetic alloys and the Slater-Pauling curve. Phys Rev B. 2009;79:115109. doi:10.1103/PhysRevB.79.115109
- Rao Z, Çakır A, Özgün Ö, et al. 3d transition-metal high-entropy invar alloy developed by adjusting the valence-electron concentration. Phys Rev Mater. 2021;5:044406. doi:10.1103/PhysRevMaterials.5.044406
- GUO S, LIU CT. Phase stability in high entropy alloys: formation of solid-solution phase or amorphous phase. Prog Nat Sci Mater Int. 2011;21:433–446. doi:10.1016/S1002-0071(12)60080-X
- Poletti MG, Battezzati L. Electronic and thermodynamic criteria for the occurrence of high entropy alloys in metallic systems. Acta Mater. 2014;75:297–306. doi:10.1016/j.actamat.2014.04.033
- Santodonato LJ, Zhang Y, Feygenson M, et al. Deviation from high-entropy configurations in the atomic distributions of a multi-principal-element alloy. Nat Commun. 2015;6:5964. doi:10.1038/ncomms6964
- Troparevsky MC, Morris JR, Kent PRC, et al. Criteria for predicting the formation of single-phase high-entropy alloys. Phys Rev X. 2015;5:011401. doi:10.1103/PhysRevX.5.011041.
- Kresse G, Furthmüller J. Efficient iterative schemes forab initiototal-energy calculations using a plane-wave basis set. Phys Rev B. 1996;54:11169–11186. doi:10.1103/PhysRevB.54.11169
- Blöchl PE. Projector augmented-wave method. Phys Rev B. 1994;50:17953–17979. doi:10.1103/PhysRevB.50.17953
- Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77:3865–3868. doi:10.1103/PhysRevLett.77.3865
- Harrison WA. Electronic structure and the properties of solids. New York: Dover Publications; 1980.
- Turchanin MA, Agraval PG. Cohesive energy, properties, and formation energy of transition metal alloys. Powder Metall Met Ceram. 2008;47:26–39. doi:10.1007/s11106-008-0006-3
- Chen W, Ding X, Feng Y, et al. Vacancy formation enthalpies of high-entropy FeCoCrNi alloy via first-principles calculations and possible implications to its superior radiation tolerance. J Mater Sci Technol. 2018;34:355–364. doi:10.1016/j.jmst.2017.11.005
- Guan H, Huang S, Ding J, et al. Chemical environment and magnetic moment effects on point defect formations in CoCrNi-based concentrated solid-solution alloys. Acta Mater. 2020;187:122–134. doi:10.1016/j.actamat.2020.01.044
- Shimizu M. Itinerant electron magnetism. Rep Prog Phys. 1981;44:329–409. doi:10.1088/0034-4885/44/4/001
- Becke AD, Edgecombe KE. A simple measure of electron localization in atomic and molecular systems. J Chem Phys. 1990;92:5397–5403. doi:10.1063/1.458517
- Henkelman G, Arnaldsson A, Jónsson H. A fast and robust algorithm for Bader decomposition of charge density. Comput Mater Sci. 2006;36:354–360. doi:10.1016/j.commatsci.2005.04.010
- Zuo T, Gao MC, Ouyang L, et al. Tailoring magnetic behavior of CoFeMnNiX (X = Al, Cr, Ga, and Sn) high entropy alloys by metal doping. Acta Mater. 2017;130:10–18. doi:10.1016/j.actamat.2017.03.013
- Marshal A, Pradeep KG, Music D, et al. Combinatorial evaluation of phase formation and magnetic properties of FeMnCoCrAl high entropy alloy thin film library. Sci Rep. 2019;9:7864. doi:10.1038/s41598-019-44351-8
- Chen S, Wang T, Li X, et al. Short-range ordering and its impact on thermodynamic property of high-entropy alloys. Acta Mater. 2022;238:118201. doi:10.1016/j.actamat.2022.118201