
ABSTRACT
Lamin B Receptor (LBR) is an inner nuclear membrane protein associated with the rare human diseases Pelger-Huët anomaly and Greenberg skeletal dysplasia. A new study has used CRISPR/Cas9-mediated genetic manipulations in a human cell system to determine that the molecular etiology of these previously poorly understood disorders is a defect in cholesterol synthesis due to loss of LBR-associated sterol C14 reductase activity. The study furthermore determined that disease-associated LBR point mutations reduce sterol C14 reductase activity by decreasing the affinity of LBR for the reducing agent NADPH. Moreover, two disease-associated LBR truncation mutants were found to be highly unstable at the protein level and are rapidly turned over by a novel nuclear membrane-based protein quality control pathway. Thus, truncated LBR variants can now be used as model substrates for further investigations of nuclear protein quality control to uncover possible implications for other disease-associated nuclear envelopathies.
Introduction
Lamin B Receptor (LBR) is a bi-functional polytopic membrane protein of the inner nuclear membrane (INM).Citation1,2 The N-terminus of LBR resides inside the nucleus and directly interacts with both chromatin and the nuclear lamina, the meshwork of intermediate filaments that underlies and supports the inner nuclear membrane ().Citation2-5 This is the function for which the protein is named, but LBR has a second, seemingly unrelated role in sterol metabolism as the C-terminal, multi-pass transmembrane domain of the protein is a functional C14 sterol reductase.Citation6,7 This second function of LBR has until now been somewhat puzzling as human cells contain another C14 sterol reductase called TM7SF2 or DHCR14 which, like most other enzymes in the cellular sterol biogenesis pathway, resides in the ER membranes and is responsive to the sterol regulatory element-binding protein (SREBP) pathway.Citation8-10 LBR, conversely, is constitutively expressed, unresponsive to the SREBP pathway, and resides instead at the INM.Citation8,11 Thus it has long been assumed that TM7SF2/DHCR14 is the primary cellular C14 sterol reductase while LBR provides at most redundant or cell type/developmental stage specific sterol biogenesis activity. However, a recent study by Tsai et al. has demonstrated not only that the sterol reductase activity of LBR is strictly required for viability under sterol-restrictive growth conditions in several human cell types, but also that two rare human diseases associated with mutations in LBR, Pelger-Huët anomaly and Greenberg skeletal dysplasia, are caused by the loss of LBR-based sterol reductase activity.Citation11
Figure 1. Localization and topology of LBR and Emerin at the inner nuclear membrane. Disease-associated LBR point mutations N547D and R583Q are indicated by filled circles and frameshift mutations by a Δ symbol at the point of truncation. (INM) inner nuclear membrane, (ONM) outer nuclear membrane, (PNS) perinuclear space.
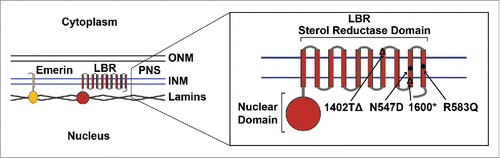
Lamin B receptor and human disease
Mutations to proteins of the nuclear lamina are known to cause a wide spectrum of human genetic diseases with phenotypes including muscular dystrophy, lipodystrophy, cardiomyopathy and the premature aging disorder Hutchinson-Gilford progeria syndrome.Citation12-15 Collectively, these disorders are known as nuclear laminopathies, but in most cases the molecular pathways by which particular laminopathic mutations manifest as specific disease phenotypes remain to be determined. Two known human diseases are associated with mutations in LBR: Pelger-Huët anomaly and Greenberg skeletal dysplasia ().Citation16-19 Pelger-Huët anomaly is a relatively benign autosomal dominant disorder characterized by hypolobulation of the normally hyperlobulated nuclei of granulocytes, a type of white blood cell.Citation19 Although studies of cells derived from the partial LBR knockout ic/ic mouse model have demonstrated a significant defect in the growth and derivation of myeloid progenitors, the multipotent cells from which all bone marrow-based blood cells are derived,Citation20 individuals displaying Pelger-Huët anomaly in granulocytes are otherwise asymptomatic. Conversely, Greenberg skeletal dysplasia (also known as hydrops-ectopic calcification-moth-eaten or HEM skeletal dysplasia) is a perinatally lethal autosomal recessive condition characterized by highly abnormal bone development (with abnormal calcification and a “moth eaten” appearance) and excessive fluid accumulation (fetal hydrops).Citation18,21-25 Interestingly, some LBR mutations found in the heterozygous state in individuals displaying Pelger-Huët anomaly have also been found in HEM/Greenberg dysplasia fetuses, and Pelger-Huët anomaly has been observed in Greenberg skeletal dysplasia parents, suggesting that at least in some cases, the two disorders represent different allelic states of the same genetic lesion, rather than distinct and unrelated genetic diseases.Citation18,21,26 As LBR is a bi-functional protein, with both nuclear lamina/chromatin binding and C14 sterol reductase activities, there has been some debate as to whether these disorders should be classified as nuclear laminopathies or as diseases of cholesterol metabolism.Citation22,27 Notably, elevated levels of the direct upstream cholesterol intermediate acted upon by the C14 sterol reductases LBR and TM7SF2 were detected in a Greenberg dysplasia fetus,Citation22 raising the question of why a deficiency in LBR is not compensated for by the putative primary C14 sterol reductase TM7SF2, in which no defect was detected. Thus the molecular etiology of these related disorders has until now remained unclear.
Table 1. Congenital diseases associated with Lamin B receptor.
Pelger-Huët anomaly and Greenberg skeletal dysplasia are loss-of-function diseases of cholesterol metabolism
Using a combination of CRISPR/Cas9 mediated knockout and Flp/Frt recombination-based gene integration, we have established a system for conducting classical genetics in a human cell system and revealed the molecular basis of the rare human diseases Pelger-Huët anomaly and Greenberg skeletal dysplasia.Citation11 We determined that LBR is required for cell viability under sterol-restrictive growth conditions in 3 different human cell lines and directly detected a decrease in cellular cholesterol content in LBR knockout cells, challenging the assumption that the C14 sterol reductase TM7SF2/DHCR14 is a redundant enzymatic activity. Interestingly, we also found no defect or change in the structure of the nuclear lamina in an LBR knockout cell line, arguing against a primarily structural role of LBR at the INM, albeit only in a tissue culture cell system.
Furthermore, we employed Flp/Frt based recombinationCitation28 to re-introduce both wild-type and disease-associated LBR alleles into an LBR knockout cell background, achieving moderate protein expression under doxycycline control and allowing for a careful analysis of both genotype and phenotype. Using this system, we determined that re-integration of wild-type LBR fully rescues the observed cholesterol auxotrophy phenotype, but that 4 separate LBR alleles associated with Pelger-Huët anomaly and Greenberg skeletal dysplasia ()() do not.
Further investigation of these four disease-associated LBR alleles revealed two separate and distinct mechanisms by which LBR mutation leads to a defect in cholesterol production. By comparison to a published structure of a related bacterial sterol C14 reductase,Citation29 we found that the LBR point mutations LBR N547D and LBR R583Q map to the NADPH binding pocket. As NADPH is the essential cofactor used by C14 sterol reductases to convert a carbon-carbon double bond in the cholesterol intermediate 4,4-dimethyl-5α-cholesta-8,14,24-trien-3β-ol to a single bond, any mutation which decreases the binding affinity of the enzyme for NADPH is expected to significantly decrease its reductive capacity. Indeed, we found that LBR R583Q has an approximately 7-fold decrease in affinity for NADPH compared to wild-type LBR, while LBR N547D showed an almost 12-fold decrease in NADPH binding capacity, resulting in a near-complete loss of de novo cholesterol synthesis.Citation11
Conversely, the two LBR truncation mutants, both of which result in the omission of several transmembrane helices from the C-terminus of the protein (), are highly unstable and are rapidly turned over by cellular protein quality control machinery, resulting in an extremely low steady-state protein expression level. Remarkably, it appears that truncated LBR is degraded by a mechanism that resembles the traditional ER-associated protein degradation (ERAD) pathway in that LBR ubiquitylation, a functional proteasome, and a functional p97 motor are requiredCitation30,31 (). However, unlike in the canonical ERAD pathway, which has until now only been found in the ER membranes, truncated LBR is first trafficked to the nuclear membranes before being dislocated directly into the nucleoplasm, where it is presumably degraded by nuclear proteasomes. This is, to our knowledge, the first instance in which an INM-based ERAD-like pathway has been observed in higher eukaryotes, and represents a second molecular mechanism by which LBR-associated disease mutations result in a loss of sterol C14 reductase functionality.
Figure 2. Emerging model of nuclear membrane-associated protein quality control. Truncated LBR is trafficked to the inner nuclear membrane, ubiquitylated, and dislocated into the nucleoplasm via a p97-dependent mechanism (note that p97 cofactors were omitted for clarity). Dislocated LBR is then degraded by nuclear proteasomes. Laminopathy-associated alleles may exert proteotoxic effects on nuclear protein quality control by competing for limited components of the degradation machinery or by inhibiting the pathway at the stage of ubiquitylation, INM dislocation, or proteasomal degradation. (INM) inner nuclear membrane, (ONM) outer nuclear membrane, (PNS) perinuclear space.
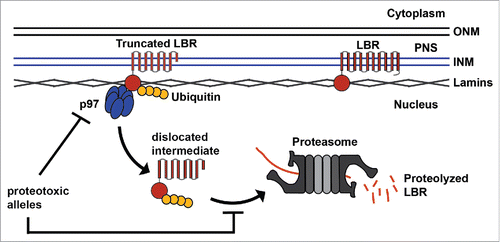
Nuclear protein quality control: Implications for other laminopathies
Having established a model system for the investigation of protein turnover at the INM, other human diseases resulting from mutations in proteins of the nuclear envelope or laminaCitation32-34 can now be examined from the perspective of protein quality control. We have previously proposed that some of these mutations may act as dominant-negative disease alleles, for example by exerting a proteotoxic effect causing aberrant protein accumulation.Citation35 Since suitable readouts for monitoring the turnover of INM proteins are now firmly established,Citation11 future studies could now determine if the molecular pathology of other laminopathy-associated alleles is related to a dominant negative overburdening or “poisoning” of the nuclear protein quality system, which would impede the otherwise timely removal of unrelated defective or otherwise undesirable proteins from the nucleoplasm or nuclear membranes (). This putative proteotoxic effect may be analogous to established proteotoxic mechanisms found in neurodegenerative diseases.Citation36,37 It is also tempting to speculate that INM-associated protein turnover machinery plays an important role beyond protein quality control, for example in regulatory proteolysis, as exemplified by INM-resident ubiquitin ligases in yeast which are involved in the regulation of sterol synthesis.Citation38,39 Additionally, it will be interesting to test whether human disease mutations which result in reduced levels of INM proteins can be explained by their degradation in the nuclear compartment, as was observed for truncated LBR.Citation11 In conclusion, a better understanding of the nuclear quality control network and its relationship to nuclear envelopathies and laminopathies is urgently required to facilitate the development of novel therapeutic avenues for the treatment of this collection of human genetic diseases.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
We thank members of the Schlieker laboratory for critically reading the manuscript and NIH for funding (1R01GM114401-01A1 to CS).
References
- Olins AL, Rhodes G, Welch DB, Zwerger M, Olins DE. Lamin B receptor: multi-tasking at the nuclear envelope. Nucleus 2010; 1:53-70; PMID:21327105; http://dx.doi.org/10.4161/nucl.1.1.10515
- Worman HJ, Evans CD, Blobel G. The lamin B receptor of the nuclear envelope inner membrane: a polytopic protein with eight potential transmembrane domains. J Cell Biol 1990; 111:1535-42; PMID:2170422; http://dx.doi.org/10.1083/jcb.111.4.1535
- Worman HJ, Yuan J, Blobel G, Georgatos SD. A lamin B receptor in the nuclear envelope. Proc Natl Acad Sci U S A 1988; 85:8531-4; PMID:2847165; http://dx.doi.org/10.1073/pnas.85.22.8531
- Pyrpasopoulou A, Meier J, Maison C, Simos G, Georgatos SD. The lamin B receptor (LBR) provides essential chromatin docking sites at the nuclear envelope. EMBO J 1996; 15:7108-19; PMID:9003786
- Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, Wilson KL. The nuclear lamina comes of age. Nat Rev Mol Cell Biol 2005; 6:21-31; PMID:15688064; http://dx.doi.org/10.1038/nrm1550
- Holmer L, Pezhman A, Worman HJ. The Human Lamin B Receptor/Sterol Reductase Multigene Family. Genomics 1998; 54:469-76; PMID:9878250; http://dx.doi.org/10.1006/geno.1998.5615
- Silve S, Dupuy PH, Ferrara P, Loison G. Human lamin B receptor exhibits sterol C14-reductase activity in Saccharomyces cerevisiae. Biochim Biophys Acta 1998; 1392:233-44; http://dx.doi.org/10.1016/S0005-2760(98)00041-1
- Bennati AM, Castelli M, Della Fazia MA, Beccari T, Caruso D, Servillo G, Roberti R. Sterol dependent regulation of human TM7SF2 gene expression: Role of the encoded 3β-hydroxysterol Δ14-reductase in human cholesterol biosynthesis. Biochim Biophys Acta 2006; 1761:677-85; http://dx.doi.org/10.1016/j.bbalip.2006.05.004
- Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997; 89:331-40; PMID:9150132; http://dx.doi.org/10.1016/S0092-8674(00)80213-5
- Sharpe LJ, Brown AJ. Controlling Cholesterol Synthesis beyond 3-Hydroxy-3-methylglutaryl-CoA Reductase (HMGCR). J Biol Chem 2013; 288:18707-15; PMID:23696639; http://dx.doi.org/10.1074/jbc.R113.479808
- Tsai PL, Zhao C, Turner E, Schlieker CD. The Lamin B receptor is essential for cholesterol synthesis and perturbed by disease-causing mutations. eLife 2016; 5:e16011; PMID:27336722
- Burke B, Stewart CL. The Laminopathies: The Functional Architecture of the Nucleus and Its Contribution to Disease*. Annu Rev Genomics Hum Genet 2006; 7:369-405; PMID:16824021; http://dx.doi.org/10.1146/annurev.genom.7.080505.115732
- Somech R, Shaklai S, Amariglio N, Rechavi G, Simon AJ. Nuclear envelopathies–raising the nuclear veil. Pediatr Res 2005; 57:8R-15R; PMID:15817509; http://dx.doi.org/10.1203/01.PDR.0000159566.54287.6C
- Worman HJ, Bonne G. “Laminopathies:” a wide spectrum of human diseases. Exp Cell Res 2007; 313:2121-33; PMID:17467691; http://dx.doi.org/10.1016/j.yexcr.2007.03.028
- Worman HJ, Ostlund C, Wang Y. Diseases of the nuclear envelope. Cold Spring Harb Perspect Biol 2010; 2:a000760; PMID:20182615; http://dx.doi.org/10.1101/cshperspect.a000760
- Hoffmann K, Dreger CK, Olins AL, Olins DE, Shultz LD, Lucke B, Karl H, Kaps R, Müller D, Vayá A, et al. Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger-Huët anomaly). Nat Genet 2002; 31:410-4; PMID:12118250
- Shultz LD, Lyons BL, Burzenski LM, Gott B, Samuels R, Schweitzer PA, Dreger C, Herrmann H, Kalscheuer V, Olins AL, et al. Mutations at the mouse ichthyosis locus are within the lamin B receptor gene: a single gene model for human Pelger–Huët anomaly. Hum Mol Genet 2003; 12:61-9; PMID:12490533; http://dx.doi.org/10.1093/hmg/ddg003
- Konstantinidou A, Karadimas C, Waterham HR, Superti-Furga A, Kaminopetros P, Grigoriadou M, Kokotas H, Agrogiannis G, Giannoulia-Karantana A, Patsouris E, et al. Pathologic, radiographic and molecular findings in three fetuses diagnosed with HEM/Greenberg skeletal dysplasia. Prenat Diagn 2008; 28:309-12; PMID:18382993; http://dx.doi.org/10.1002/pd.1976
- Best S, Salvati F, Kallo J, Garner C, Height S, Thein SL, Rees DC. Lamin B-receptor mutations in Pelger–Huët anomaly. Br J Haematol 2003; 123:542-4; PMID:14617022; http://dx.doi.org/10.1046/j.1365-2141.2003.04621.x
- Subramanian G, Chaudhury P, Malu K, Fowler S, Manmode R, Gotur D, Zwerger M, Ryan D, Roberti R, Gaines P. Lamin B Receptor Regulates the Growth and Maturation of Myeloid Progenitors via its Sterol Reductase Domain: Implications for Cholesterol Biosynthesis in Regulating Myelopoiesis. J Immunol 2012; 188:85-102; PMID:22140257; http://dx.doi.org/10.4049/jimmunol.1003804
- Oosterwijk JC, Mansour S, van Noort G, Waterham HR, Hall CM, Hennekam RCM. Congenital abnormalities reported in Pelger-Huët homozygosity as compared to Greenberg/HEM dysplasia: highly variable expression of allelic phenotypes. J Med Genet 2003; 40:937-41; PMID:14684694; http://dx.doi.org/10.1136/jmg.40.12.937
- Waterham HR, Koster J, Mooyer P, Noort Gv, Kelley RI, Wilcox WR, Wanders RJ, Hennekam RC, Oosterwijk JC. Autosomal Recessive HEM/Greenberg skeletal dysplasia Is Caused by 3β-Hydroxysterol Δ14-Reductase Deficiency Due to Mutations in the Lamin B Receptor Gene. Am J Hum Genet 2003; 72:1013-7; PMID:12618959; http://dx.doi.org/10.1086/373938
- Chitayat D, Gruber H, Mullen BJ, Pauzner D, Costa T, Lachman R, Rimoin DL. Hydrops‐ectopic calcification—moth‐eaten skeletal dysplasia (Greenberg dysplasia): Prenatal diagnosis and further delineation of a rare genetic disorder. Am J Med Genet 1993; 47:272-7; PMID:8213919; http://dx.doi.org/10.1002/ajmg.1320470226
- Horn LC, Faber R, Meiner A, Piskazeck U, Spranger J. Greenberg dysplasia: first reported case with additional non‐skeletal malformations and without consanguinity. Prenat Diagn 2000; 20:1008-11; PMID:11113916; http://dx.doi.org/10.1002/1097-0223(200012)20:12%3c1008::AID-PD954%3e3.0.CO;2-S
- Greenberg CR, Rimoin DL, Gruber HE, DeSa D, Reed M, Lachman RS. A new autosomal recessive lethal chondrodystrophy with congenital hydrops. Am J Med Genet 1988; 29:623-32; PMID:3377005; http://dx.doi.org/10.1002/ajmg.1320290321
- Clayton P, Fischer B, Mann A, Mansour S, Rossier E, Veen M, Lang C, Baasanjav S, Kieslich M, Brossuleit K, et al. Mutations causing Greenberg dysplasia but not Pelger anomaly uncouple enzymatic from structural functions of a nuclear membrane protein. Nucleus 2010; 1:354-66; PMID:21327084; http://dx.doi.org/10.4161/nucl.1.4.12435
- Wassif CA, Brownson KE, Sterner AL, Forlino A, Zerfas PM, Wilson WK, Starost MF, Porter FD. HEM dysplasia and ichthyosis are likely laminopathies and not due to 3β-hydroxysterol Δ14-reductase deficiency. Hum Mol Genet 2007; 16:1176-87; PMID:17403717; http://dx.doi.org/10.1093/hmg/ddm065
- Turner EM, Brown RS, Laudermilch E, Tsai PL, Schlieker C. The Torsin Activator LULL1 Is Required for Efficient Growth of Herpes Simplex Virus 1. J Virol 2015; 89:8444-52; PMID:26041288; http://dx.doi.org/10.1128/JVI.01143-15
- Li X, Roberti R, Blobel G. Structure of an integral membrane sterol reductase from Methylomicrobium alcaliphilum. Nature 2015; 517:104-7; PMID:25307054; http://dx.doi.org/10.1038/nature13797
- Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol 2008; 9:944-57; PMID:19002207; http://dx.doi.org/10.1038/nrm2546
- Zattas D, Hochstrasser M. Ubiquitin-dependent protein degradation at the yeast endoplasmic reticulum and nuclear envelope. Crit Rev Biochem Mol Biol 2015; 50:1-17; PMID:25231236; http://dx.doi.org/10.3109/10409238.2014.959889
- Dauer WT, Worman HJ. The nuclear envelope as a signaling node in development and disease. Dev Cell 2009; 17:626-38; PMID:19922868; http://dx.doi.org/10.1016/j.devcel.2009.10.016
- Burke B, Stewart CL. Functional architecture of the cell's nucleus in development, aging, and disease. Curr Topics Dev Biol 2014; 109:1-52; PMID:24947235; http://dx.doi.org/10.1016/B978-0-12-397920-9.00006-8
- Gruenbaum Y, Foisner R. Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu Rev Biochem 2015; 84:131-64; PMID:25747401; http://dx.doi.org/10.1146/annurev-biochem-060614-034115
- Rose A, Schlieker C. Alternative nuclear transport for cellular protein quality control. Trends Cell Biol 2012; 22:509-14; PMID:22858153; http://dx.doi.org/10.1016/j.tcb.2012.07.003
- Labbadia J, Morimoto RI. Huntington's disease: underlying molecular mechanisms and emerging concepts. Trends Biochem Sci 2013; 38:378-85; PMID:23768628; http://dx.doi.org/10.1016/j.tibs.2013.05.003
- Balchin D, Hayer-Hartl M, Hartl FU. In vivo aspects of protein folding and quality control. Science 2016; 353:aac4354; PMID:27365453; http://dx.doi.org/10.1126/science.aac4354
- Khmelinskii A, Blaszczak E, Pantazopoulou M, Fischer B, Omnus DJ, Le Dez G, Brossard A, Gunnarsson A, Barry JD, Meurer M, et al. Protein quality control at the inner nuclear membrane. Nature 2014; 516:410-3; PMID:25519137; http://dx.doi.org/10.1038/nature14096
- Foresti O, Rodriguez-Vaello V, Funaya C, Carvalho P. Quality control of inner nuclear membrane proteins by the Asi complex. Science 2014; 346:751-5; PMID:25236469; http://dx.doi.org/10.1126/science.1255638