
Abstract
Background: Amyotrophic lateral sclerosis (ALS) is a progressive fatal disease with a varying range of clinical characteristics. Objective: To describe the clinical characteristics in a large cohort of ALS participants enrolled in the National ALS Registry. Methods: Data from ALS participants who completed the Registry’s online clinical survey module during 2010–2015 were analyzed to determine characteristics, such as site of onset, associated symptoms, time of symptom onset to diagnosis, time of diagnosis to hospice referral, and pharmacological and non-pharmacological interventions. Results: Of the 1758 participants who completed the survey, 60.9% were male, 62.1% were 50–69 years old, and 95.5% white. Approximately, 72.0% reported initial limb weakness onset of disease, followed by bulbar (22.1%), and trunk/global onset (6.1%). Other symptoms ever experienced included cramps (56.7%), fasciculations (56.3%), and dysarthria (33.0%). The median time between an increase of muscle cramps until an ALS diagnosis was 12 months; limb onset participants had cramps longer preceding diagnosis versus those with bulbar onset. The most frequent interventions used included riluzole (48.3% currently using), wheelchairs/scooters (32.8%), and noninvasive breathing equipment (30.0%). Participants with trunk/global onset were referred to hospice almost four times earlier than others. Conclusions: These data show how ALS clinical characteristics differ widely in a large cohort of participants preceding diagnosis and reflect variations in disease onset, progression, and prognosis. Better characterization of symptom onset may assist clinicians in diagnosing ALS sooner, which could lead to earlier therapeutic interventions.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive, fatal neurodegenerative disorder defined by the loss of upper and lower motor neurons, typically resulting in death within 2–5 years of initial symptom onset (Citation1). Despite ALS being defined in 1869 (Citation2), the actual pathogenesis and cause(s) remain largely unknown and there is currently no cure. Conservative estimates suggest that in the United States (US) over 16,000, or 5.2/100,000 people, lived with ALS in 2015 (Citation3) and approximately 5000, or 1.5/100,000, are diagnosed annually (Citation4).
While death due to ventilatory failure from ALS affects most with the disease, there is a wide range of clinical characteristics among patients (e.g. site of onset and progression rate) (Citation5). Genetically determined ALS accounts for about 10% of cases, whereas the remaining cases are considered sporadic (Citation6). Additionally, site of onset results in several different phenotypes that include limb-onset, bulbar-onset, or trunk/global onset. Those with bulbar onset typically have shorter life expectancy versus those with limb onset (Citation7). Furthermore, patients may experience various symptoms during the course of the disease, such as muscle cramps, fasciculations, dysarthria, and dysphagia (Citation8).
The purpose of this article is to describe the clinical characteristics of a large cohort of US participants enrolled in the National ALS Registry. Since ALS onset and progression varies greatly by patient, these data provide additional information on phenotypic differences in a national population (Citation9,Citation10). Having a better understanding of ALS onset may assist clinicians in making a quicker diagnosis, which could lead to earlier therapeutic interventions.
Methods
The National ALS Registry
In October 2010, the US federal Agency for Toxic Substances and Disease Registry (ATSDR), part of the Centers for Disease Control and Prevention (CDC), launched the congressionally mandated, population-based National ALS Registry (Registry) to help clarify the epidemiology of ALS in the US (Citation11). While details about the Registry’s objectives are presented elsewhere (Citation3), briefly, the Registry’s purpose is to quantify the incidence and prevalence of ALS in the US, describe the patient demographics, and examine potential risk factors (Citation12). Similarly, the Registry’s methods also have been previously described (Citation13). Cases from both the national administrative databases and the web portal are merged and de-duplicated to ensure that individuals are not counted twice. To verify ALS status within the web portal, ATSDR adopted the six questions from the U.S. Department of Veterans Affairs ALS registry that has been proven to be reliable indicators for accurate ALS diagnoses (Citation14).
The Registry’s web portal also allows participants to complete brief online surveys about their ALS experience on topics such as occupational and military histories, smoking and alcohol use, and clinical symptoms. Currently, there are 17 survey modules available shown in Supplementary Table 1. These surveys were designed by Stanford University (Citation15) and are structured such that participants can answer the questions without having to involve a healthcare provider. Due to the nature of these online surveys, it is likely there is a self-selection bias within the study. These data are likely slanted toward a younger and better educated patient sample. To-date, over 80,000 surveys have been completed representing the largest, most geographically diverse collection of ALS risk factor data available.
Clinical symptoms survey module
The clinical symptoms survey module was created in partnership with the ALS Research Group, which was made up of US and Canadian neurologists and researchers. The purpose of the module is to examine physical symptoms participants developed before and after an ALS diagnosis. The survey contains 54 questions and covers topics such as site of onset, time of initial symptom onset to diagnosis, and time of diagnosis to hospice referral as shown in Supplementary Table 2. This module launched in December 2013 to new Registry enrollees and previous enrollees were prompted to return to the web portal to complete this survey. Therefore, this analysis covers from 19 October 2010 to 31 December 2015.
Data analysis
Selected demographic characteristics for those who completed the clinical survey module were abstracted including gender, race, ethnicity, age at diagnosis, as well as geographic region of residence at the time of diagnosis. Race was defined by standard US definitions; a primary race was noted and if more than one race was chosen, participants were categorized as nonwhite. Geographic regions were created by combining US Department of Health and Human Services (HHS) regions Supplementary Table 2. For these groupings, the Northeastern region included HHS Regions 1, 2, and 3; Southern region included HHS Regions 4 and 6; Midwestern region included HHS Regions 5 and 7; and Western region included HHS Regions 8, 9, and 10 (Citation16).
“Site of onset” refers to the body part where a participant reported their first ALS related weakness or symptom. The body was divided into 3 groups: 1) limb—extremities included hand, arm, foot or leg, 2) bulbar—included speech and/or swallowing, and 3) trunk/global included neck, back or abdominal areas, breathing muscles, or total body weakness. “Other symptoms” were ones which participants experienced after the initial symptom and could have occurred pre or post-diagnosis. The prevalence of symptoms was determined by calculating the time from symptom onset to diagnosis. Univariate analyses were performed to examine the associations among the site of onset (limb, bulbar, or trunk/global), symptoms following initial onset, and interventions for ALS. Categorical variables were assessed with Chi-square tests. Statistical significance was considered at 0.05. All data analysis was performed using SAS version 9.4 (SAS Institute Inc., Cary, NC).
Results
Between 19 October 2010 and 31 December 2015, 6352 adults, 18 years or older, registered via the Registry’s online portal and completed at least one of the 17 surveys. Of these, 1758 (27.7%) completed the clinical survey module as well as basic demographic information. Demographic characteristics of these 1758 participants are displayed in . Over 60% of the participants were aged 50–69 years and male, and almost 96% were white. One-third of participants resided in the Southern part of the US at the time of diagnosis, closely followed by the Midwest ().
Table 1 Demographic characteristics among US adults with ALS who responded to the National ALS Registry’s Clinical Survey Module (19 October 2010–31 December 2015).
Site of onset among the 1758 participants stratified by gender is presented in . All participants reported having progressive muscle weakness prior to ALS diagnosis. Almost three-fourths of participants (n = 1261) had limb onset. A significantly higher proportion of male participants had limb onset weakness (p = 0.02) and weakness first in their hand or arm (p = 0.02). Approximately, 22% of the 1758 participants experienced bulbar onset; of those, females were statistically more likely to have bulbar onset (p = 0.002). Trunk/global onset accounted for 6.1% of participants with an even gender distribution. The study also compared gross motor tasks, fine motor tasks, bulbar functions, and respiratory functions through validated question-based scales by site of onset. This scale, the Amyotrophic Lateral Sclerosis Functional Rating Scale-revised (ALSFRS-R), measures physical function in carrying out activities of daily living of patients with ALS (Citation17). The rate of estimated change of ALSFRS scores by site of onset was examined and bulbar participants declined faster compared to other groups (data not presented). The mean age of onset was 59.1 years overall, 58.1 years for limb onset, 61.5 years for bulbar onset, and 62.6 years for trunk/global onset.
Table 2 Initial site of onset among 1758Table Footnotea US adults with ALS who responded to the National ALS Registry’s Clinical Survey Module (19 October 2010–31 December 2015).
Other symptoms experienced are presented in . The most frequent symptoms included muscle cramps (56.7%), fasciculations (56.3%), and dysarthria (33.0%). Approximately one-fourth (n = 447) had experienced trips or falls. Almost 13% of participants had difficulty controlling bowels, 6.7% had experienced pneumonia, and 3.9% experienced blood clots. When stratified by gender, a higher proportion of females reported suffering from pneumonia (p = 0.04) as well as having problems with speech (p = 0.02).
Table 3 Other symptoms experienced among 1758Table Footnotea US adults with ALS who responded to the National ALS Registry’s Clinical Survey Module (19 October 2010–31 December 2015).
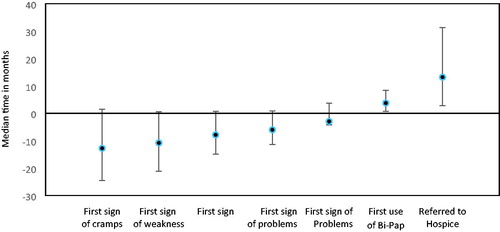
The time between symptom-onset and ALS diagnosis was calculated for each symptom (). Post muscle cramp onset, the median time to diagnosis was just over one year. Post muscle weakness onset, an ALS diagnosis was made after almost 11 months, followed by twitching at almost 8 months prior to ALS diagnosis. Of the 581 participants with speech onset, median time to ALS diagnosis was approximately six months. Bowel issues preceded an ALS diagnosis by approximately three months.
Figure 1 Median time from ALS diagnosis to symptoms onset among adults with ALS, 19 October 2010–31 December 2015.
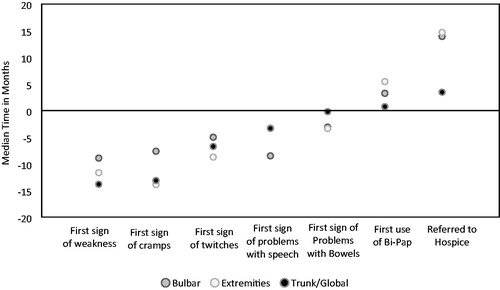
Bulbar onset participants were typically diagnosed faster than those with limb or trunk/global onset (). The average time to diagnosis for bulbar onset individuals was almost nine months, compared to almost one year for limb onset and nearly 14 months for trunk/global onset. For symptoms of cramps and twitching, bulbar onset participants again reported shorter duration of symptoms to diagnosis as compared to those with limb or trunk/global onset (7.6 and 5 months, respectively). Those with trunk/global onset had speech and bowel symptoms (3.3 and 0.2 months, respectively) for a much shorter prior to diagnosis versus those with bulbar onset (8.5 and 3.1 months, respectively) and limb onset (3.3 and 3.4 months, respectively).
Figure 2 Median time from ALS diagnosis to symptoms onset stratified on body part weakness among adults with ALS, 19 October 2010–31 December 2015.
The use of pharmacological and non-pharmacological interventions used is presented in . The most frequent interventions included riluzole, the only Food and Drug Administration approved ALS drug available during the study period (48.3% currently using), wheelchairs/scooters (32.8%), and noninvasive breathing equipment (30.0%). Noninvasive breathing equipment was more commonly started after diagnosis with a median time of three months. Sixteen percent of participants used a communication device, with females being more likely to use (p = 0.09). Only 2.6% of participants reported having a tracheotomy, with an equal split by gender. Approximately, 69% of participants had visited a multidisciplinary clinic, while 25% had been involved in ALS research studies and clinical trials, and 18.6% had genetic testing done (data for the latter two not presented in table).
Table 4 Use of Interventions among 1758Table Footnotea US Adults with ALS who responded to the National ALS Registry’s Clinical Survey Module (19 October 2010–31 December 2015).
Regarding end-of-life care, 91 participants had been referred to hospice; the timeframe from diagnosis to referral was just over 13 months. Women were referred to hospice earlier at 9.2 months and men later at 14 months. Age at diagnosis also affected time to hospice referral with older participants being referred earlier: age 18–39 at 40.5 months, 40–49 at 29 months, 50–59 at 14.2 months, 60–69 at 10.4 months, and those ≥70 years at 9.5 months. Trunk/global onset participants were referred to hospice sooner after diagnosis with a median time of 3.5 months compared with bulbar (13.9 months) and limb onset participants (14.7 months). Two-third of participants had advanced directives regarding end-of-life-care. Participants who were on, or had been on, riluzole were referred to hospice at 15 months after diagnosis and those never taking riluzole earlier at 10 months.
Discussion
This US dataset allows for an exploration of ALS symptoms on a national scale and validates findings seen in earlier studies. Initial disease onset symptoms precede an ALS diagnosis by just less than one year in several US series and in this large US series by less than 10 months (Citation18,Citation19). This diagnostic delay can lead to unnecessary diagnostic evaluations (Citation20) and delay treatments shown to be effective (Citation21,Citation22). Specific descriptions of the first symptoms of the disease could assist clinicians in making an earlier diagnosis. These data presented complement past reports (Citation23,Citation24). Some European studies have published a median time from symptom onset to an ALS diagnosis in a little as less than 8 months while others have seen a median diagnosis delay of up to 12.5 months (Citation25,Citation26).
Muscle cramps are reported in 56.7% of participants in this cohort. While this is lower than the 92–95% of patients reporting cramps during their ALS disease (Citation27,Citation28), it is higher than the 11.4% estimate by Eisen (Citation23). Muscle cramps are highly variable over time, but there is no consistent trend (Citation28). Of the almost 60% of participants who experienced muscles cramps, an ALS diagnosis came almost 13 months later.
Fasciculations are another lower motor neuron hyperexcitability phenomena that strongly associate with ALS, but for which prevalence numbers have not been described (Citation29); in this series, 56.3% report muscle twitching. Fasciculations, and rarely also cramps, can occur and go unnoticed by the patient; this may result in some underreporting of these symptoms. While muscle cramps and fasciculations are predominantly lower motor neuron symptoms, they are a feature of neuronal hyperexcitability that frequently precede weakness. The median occurrences of different symptoms prior to diagnosis as depicted in provide an overview of how symptoms develop in ALS.
Trips and falls are byproducts of ALS lower extremity involvement and 25.4% of the participants reported these, with a slightly larger number (36.9%) reporting leg or foot weakness as the initial onset symptom; this compares to prior series of 29% (Citation30) and 35% (Citation31), respectively. In this series, approximately a third of participants report utilizing a motorized wheelchair or scooter. These types of devices can reduce the risk of trips/falls and provide a way of maintaining independent mobility later in the disease.
In this study, blood clots are defined as having one in your arm, leg, or lung requiring treatment with a blood thinner. Almost 4% of the participants reported having a blood clot that required treatment. This far exceeds CDC’s estimate of 1–2 per 1000 (Citation32). This ALS study has 20 times the expected number of participants experiencing blood clots. ALS patients typically have a more sedentary lifestyle, could be protein deficit, and are typically an older population, all of which may lead to the development of blood clots.
Almost 12% of participants reported having changes in bowel movements as a symptom that preceded an ALS diagnosis by roughly three months. Recent findings reported that weight loss in ALS can precede presumed disease onset (Citation33). ALS also has some involvement of the autonomic nervous system which could result in bowel problems (Citation34).
Tracheostomy is a rare treatment choice in the US and low numbers have been reported in prior series (Citation35). In this study, 2.6% of participants had chosen this intervention. The portion of the participants with communication difficulties, making participation in the survey difficult, is likely higher in participants with a tracheostomy than in the general ALS population and could lead to underreporting of the intervention.
There were few differences by gender, but referral to hospice occurred earlier for women. This correlates with shorter survival time women experience, which could be explained by older age and more frequent bulbar onset, or could possibly be due to less aggressive management (Citation36). Time in hospice is generally short and earlier referrals are often beneficial. Older participants who also have a shorter expected survival (Citation37,Citation38) were also referred to hospice earlier. Current and past use of riluzole was associated with longer time to hospice referral. This could be due to participants choosing more aggressive treatments aimed at prolonging survival, which delay their entry into hospice, which has been seen in other registry studies (Citation39,Citation40). All study participants were alive so we were unable to estimate any differences in survival. Riluzole use in the US continues to be low (48% in the Registry) compared to other industrialized nations, such as Sweden at 76% and Japan at 58% (Citation41,Citation42).
Another difference among genders was site of onset. Women were more likely to have bulbar onset. Of the women in this study, almost 26% had bulbar onset, for men the proportion was just under 20% , while just over 22% of all participants had bulbar site of onset. This is similar or slightly lower than other studies finding between 22 and almost 41% (Citation27,Citation28). However, most of these studies do have different case identification schemes.
A major study limitation is that the survey is not a random sample from the database, and it is likely that some biases are introduced by the self-identification process. Other studies utilize the provision of free or subsidized medicine regardless of age or socio-economic status to identify cases (Citation26,Citation27,Citation42). Participants with internet access are presumably more likely to participate; this may skew the population toward a younger, demographically white, and better educated patient sample. The participants in this study were mostly (62.1%) between the ages of 50 and 69, which is a somewhat larger proportion than what is seen in the National ALS Registry as a whole (48.2%) (Citation3), and clearly contrasting with the overall U.S. population in this segment (24.4%) (Citation43). The portion of younger participants is over-represented in this sample and the oldest age group under represented as compared to the Registry as a whole (). While the survey participants’ spatial distribution is comparable to the overall Registry, racial diversity appears to be underrepresented in the sample with only 4.5% being nonwhite as compared to 12.1% in the registry as a whole (Citation3,Citation41). Potential causes include lower access to computers that are required for self-registration; reduced awareness of the Registry perhaps due to lower use of ALS specialty clinics; and reduced participation by western residents, a region comprising a substantial nonwhite population (Citation44). Another possible study limitation is recall bias. Participants were asked to enter dates and symptoms before they were diagnosed with ALS. It is possible participants estimated the time from the particular symptom to an ALS diagnosis if they did not remember the exact date each symptom started. However, answering surveys is voluntary and not everyone who registered took this survey. A final limitation is duplication of participants. While every attempt is taken to include only unique registrants in our analyses, there remains the possibility that duplicate registrations could be included. Conversely, additional analyses were taken to review the data for duplication and the analysis indicated that very few duplicates likely occurred.
Conclusion
An ALS diagnosis is primarily based on clinical assessment; therefore, it is not surprising that a diagnosis is often deferred to specialists with expertise in this rare condition after several months of testing. The Registry data presented here show how ALS symptoms vary in participants before they are diagnosed and reflect current published findings on disease onset. This study is largely consistent with results of other reported research on smaller, less geographically diverse populations. Better characterization of symptom onset can assist clinicians in making referrals to an ALS specialist resulting in earlier diagnosis, which could lead to earlier therapeutic interventions. Further research into the early symptoms of ALS is still needed in order to better understand disease onset.
Declaration of interest
The CDC/ATSDR authors have no declarations of interest. Dr. Oskarsson has no declarations of interest linked to the study. He has served as a consultant for Biogen and FlexPharma in 2018 and receives research funding from Cytokinetics, Biogen, Orion, Eisai, and Roche.
Disclaimer
The findings and conclusions in this report are those of the authors and do not necessarily represent the official positions of the Agency for Toxic Substances and Disease Registry, the Centers for Disease Control and Prevention, and/or the US Department of HHS.
Supplementary_Table_2.docx
Download MS Word (26.8 KB)Supplementary_Table_1.docx
Download MS Word (24.3 KB)Acknowledgments
The authors are grateful to those living with ALS who give their valuable time to contribute important health data to researchers. Without their help, these findings, and countless others, would not be possible.
Additional information
Funding
Related Research Data
References
- Mitsumoto HCD, Pioro EP. Amyotrophic lateral sclerosis. Philadelphia, PA: F.A. Davis Company; 1998.
- Charcot JMJ. A. Deux cas d’atrophie musculaire progressive avec lesions de la substance grise et des faisceaux antero-lateraux de la moelle epiniere [French]. Arch Physiol Neurol Pathol. 1869;2:744.
- Mehta P, Kaye W, Raymond J, Punjani R, Larson T, Cohen J. Prevalence of amyotrophic lateral sclerosis — United States, 2015. Morb Mortal Wkly Rep. 2018;67:1285–9. https://www.cdc.gov/mmwr/volumes/67/wr/pdfs/mm6746a1-H.pdf
- Wagner L, Rechtman L, Jordan H, Ritsick M, Sanchez M, Sorenson E, Kaye W. State and metropolitan area-based amyotrophic lateral sclerosis (ALS) surveillance. Amyotroph Lateral Scler Frontotemporal Degener. 2015;17:128–34.
- Ravits J, Appel S, Baloh RH, Barohn R, Brooks BR, Elman L, et al. Deciphering amyotrophic lateral sclerosis: what phenotype, neuropathology, and genetics are telling us about pathogenesis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:5–18.
- Boylan K. Familial amyotrophic lateral sclerosis. Neurol Clin. 2015;33:807–30.
- Chiò A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler. 2009;10:310–23.
- Ringel SP, Murphy JR, Alderson MK, Bryan W, England JD, Miller RG, et al. The natural history of amyotrophic lateral sclerosis. Neurology. 1993;43:1316–22.
- Chiò A, Calvo A, Mazzini L, Cantello R, Mora G, Moglia C, et al. Extensive genetics of ALS: a population-based study in Italy. Neurology. 2012;79:1983–9.
- Al-Chalabi A, Hardiman O, Kiernan MC, Chiò A, Rix-Brooks B, van den Berg LH. Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol. 2016;15:1182–94.
- US Public Health Service. ALS registry act. 110th congress. Public law. Washington, DC: US Public Health Service, 2008:110–373. 122 Stat 4047. Available at: http://wwwn.cdc.gov/als/Download/ALS%20Registry%20Act%20.(Public%20Law%20110-373).pdf.
- Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci. 1994;124:96–107.
- Antao VC, Horton DK. The national amyotrophic lateral sclerosis (ALS) registry. J Environ Health. 2012;75:28–30.
- Allen KD, Kasarskis EJ, Bedlack RS, Rozear MP, Morgenlander JC, Sabet A, et al. The national registry of veterans with amyotrophic lateral sclerosis. Neuroepidemiology. 2008;30:180–90. PMID: 18421218.
- Horton DK, Mehta P, Antao V. Quantifying a nonnotifiable disease in the United States: the national amyotrophic lateral sclerosis registry model. JAMA. 2014;312:1097–8.
- US Department of Health and Human Services. US department of health and human services regions. Washington, DC: US Department of Health and Human Services; 2017. Available at: https://www.hhs.gov/about/agencies/iea/regional-offices/index.html.
- Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, Nakanishi A. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. J Neurol Sci. 1999;169:13–21.
- Khishchenko N, Allen KD, Coffman CJ, Kasarskis EJ, Lindquist JH, Morgenlander JC, et al. Time to diagnosis in the national registry of veterans with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010;11:125–32.
- Paganoni S, Macklin EA, Lee A, Murphy A, Chang J, Zipf A, et al. Diagnostic timelines and delays in diagnosing amyotrophic lateral sclerosis (ALS). Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:453–6.
- Shook SJ, Pioro EP. Racing against the clock: recognizing, differentiating, diagnosing, and referring the amyotrophic lateral sclerosis patient. Ann Neurol. 2009;65:S10–S6.
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/riluzole study group. N Engl J Med. 1994;330:585–91.
- Abe K, Aoki M, Tsuji S, Itoyama Y, Sobue G, Togo M, et al. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017;16:505–12.
- Eisen A. How to improve the diagnostic process. J Neurol. 1999;246:III6–9.
- Galvin M, Gaffney R, Corr B, Mays I, Hardiman O. From first symptoms to diagnosis of amyotrophic lateral sclerosis: perspectives of an Irish informal caregiver cohort-a thematic analysis. BMJ Open. 2017;7:e014985.
- Logroscino G, Traynor BJ, Hardiman O, Chio A, Mitchell D, Swingler RJ, et al. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry. 2010;81:385–90.
- Hardiman O, Al-Chalabi AA, Brayne C, Beghi E, van de Berg LH, Chio A, et al. The changing picture of amyotrophic lateral sclerosis: lessons from European registers. J Neurol Neurosurg Psychiatry. 2017;88:557–63.
- Stephens HE, Joyce NC, Oskarsson B. National study of muscle cramps in ALS in the USA. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:32–36.
- Caress JB, Ciarlone SL, Sullivan EA, Griffin LP, Cartwright MS. Natural history of muscle cramps in amyotrophic lateral sclerosis. Muscle Nerve. 2016;53:513–7.
- Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SSW, Sandoe J, et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014;7:1–11.
- Turner MR, Brockington A, Scaber J, Hollinger H, Marsden R, Shaw PJ, et al. Pattern of spread and prognosis in lower limb-onset ALS. Amyotroph Lateral Scler. 2010;11:369–73.
- Fujimura-Kiyono C, Kimura F, Ishida S, Nakajima H, Hosokawa T, Sugino M, et al. Onset and spreading patterns of lower motor neuron involvements predict survival in sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2011;82:1244–9.
- Beckman MG, Hooper WC, Critchley SE, Ortel TL. Venous thromboembolism: a public health concern. Am J Prev Med. 2010;38:S495–S501.
- Moglia C, Calvo A, Grassano M, Canosa A, Manera U, D’Ovidio F, et al. Early weight loss in amyotrophic lateral sclerosis: outcome relevance and clinical correlates in a population-based cohort. J Neurol Neurosurg Psychiatry. 2019;0:1–8.
- Piccione EA, Sletten DM, Staff NP, Low PA. Autonomic system and amyotrophic lateral sclerosis. Muscle Nerve. 2015;51:676–9.
- Moss AH, Casey P, Stocking CB, Roos RP, Brooks BR, Siegler M. Home ventilation for amyotrophic lateral sclerosis patients: outcomes, costs, and patient, family, and physician attitudes. Neurology. 1993;43:438–43.
- del Aguila MA, Longstreth WT Jr, McGuire V, Koepsell TD, van Belle G. Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology. 2003;60:813–9.
- Traxinger K, Kelly C, Johnson BA, Lyles RH, Glass JD. Prognosis and epidemiology of amyotrophic lateral sclerosis: analysis of a clinic population, 1997–2011. Neurol Clin Pract. 2013;3:313–20.
- Chen L, Liu X, Tang L, Zhang N, Fan D. Long-term use of riluzole could improve the prognosis of sporadic amyotrophic lateral sclerosis patients: a real-world cohort study in China. Front Aging Neurosci. 2016;8:246.
- Mandrioli J, Malerba SA, Beghi E, Fini N, Fasano A, Zucchi E, et al. Riluzole and other prognostic factors in ALS: a population-based registry study in Italy. J Neurol. 2018; 265:817–27.
- Nygren I, Antonova K, Mattsson P, Askmark H. The ALS/MND prevalence in Sweden estimated by riluzole sales statistics. Acta Neurol Scand. 2005;111:180–4.
- Watanabe H, Atsuta N, Nakamura R, Hirakawa A, Watanabe H, Ito M, et al. Factors affecting longitudinal functional decline and survival in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:230–6.
- Rooney JPK, Visser AE, D’Ovidio F, Vermeulen R, Beghi E, Chio A, et al. A case-control study of hormonal exposures as etiologic factors for ALS in women. Neurology. 2017;89:1283–90.
- US Census Bureau. Total population 2014 American community survey 1-year. Washington, DC: US Department of Commerce, US Census Bureau; 2014.
- Kaye WE, Wagner L, Wu R, Mehta P. Evaluating the completeness of the national ALS registry, United States. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19:112–7.