
Abstract
Background: Primary lateral sclerosis (PLS) is a rare motor neuron disease characterized by progressive degeneration of upper motor neurons, resulting in spasticity and disability. There is, however, mounting evidence that the disease is not limited to upper motor neurons alone and that cognitive and behavioral changes within the spectrum of frontotemporal dementia (FTD) are part of the clinical phenotype. Objectives: To provide an in-depth classification of the cognitive and behavioral profiles of PLS by using the golden standard, a full neuropsychological evaluation, as well as a comprehensive behavioral assessment in a cohort of 30 cases. Results: Only 7 out of 30 PLS patients scored within normal range on all of the tests within our battery. The neuropsychological profile of PLS consists of deficits in social cognition (affective theory of mind (ToM) in particular), fluency, executive functions and memory. Using the revised Strong criteria, we could classify 57% of patients within the FTD spectrum (of which 17% had behavioral variant FTD). An additional 20% of patients had deficits which were not characteristic of FTD. Conclusions: This study confirms that PLS is not a restricted phenotype (only affecting upper motor neurons) and that behavioral and cognitive changes are common. Therefore, clinicians treating PLS patients should routinely assess cognition and behavior as part of routine care as cognitive and behavioral changes impact management, decision-making and care-giver burden. This assessment should be sensitive to the neuropsychological profile of PLS (social cognition (affective ToM in particular), fluency, executive functions and memory) and behavioral changes.
Introduction
Primary lateral sclerosis (PLS) is a rare neurodegenerative disease characterized by progressive degeneration of upper motor neurons (UMN). The clinical phenotype shows an insidious onset of symptoms in the legs, which slowly and relatively symmetrically ascends to the cervical and bulbar regions in roughly half of PLS cases. In the remaining patients the disease is characterized by prominent bulbar symptoms with a patchier pattern of progression (Citation1).
Whether PLS is a separate disease or a subtype of amyotrophic lateral sclerosis (ALS), is a longstanding topic of debate. The most recent version of the El Escorial criteria considers PLS to be a restricted phenotype of ALS, meaning it is characterized by isolated UMN degeneration (Citation2). There is, however, mounting evidence that PLS patients may also develop cognitive and behavioral changes within the spectrum of frontotemporal dementia (FTD).
We recently performed a review of the literature, in which we found that 2% of PLS patients were reported to have co-morbid FTD and in 22% of patients cognitive and/or behavioral changes that did not fulfill formal diagnostic criteria (Citation3). However, given the rare nature of PLS, all studies were small in size (often describing a single patient) and most applied simple neuropsychological screening instruments. Our meta-analysis, which we performed with the aim of constructing the neuropsychological profile of PLS, shows deficits in executive function, delayed verbal memory, psychomotor speed and fluency, but must be interpreted with caution as we found high heterogeneity between studies and large confidence intervals (Citation3).
In a subsequent study, we screened 75 PLS patients for cognitive and behavioral changes using the Edinburgh Cognitive and Behavioral ALS Screen (ECAS) (Citation4), Frontal Assessment Battery (FAB) (Citation5) and Amyotrophic Lateral Sclerosis-Frontotemporal Dementia-Questionnaire (ALS-FTD-Q) (Citation6). In this cohort 10% of PLS patients were found to have behavioral variant FTD (bvFTD) and deficits within the FTD-spectrum were found in approximately 30% of cases (Citation7).
Overall it appears that changes within the FTD-spectrum are a common and important feature of PLS, that requires further investigation. In this study, we, therefore, aim to provide an in-depth characterization of the cognitive and behavioral profile of PLS in a large cohort using the golden standard (full neuropsychological evaluation (NPE)) as well as a comprehensive behavioral assessment. Additionally, we examined whether the extent of cognitive and behavioral impairment could be related to disease severity and disease duration.
Material and methods
Participants
Patients with PLS were recruited between March 2016 and July 2016 from the outpatient clinic of the University Medical Center Utrecht. Both incident and prevalent cases were eligible. All patients were required to fulfill the criteria for PLS (Citation8,Citation9), able to communicate verbally and able to provide informed consent. Disease severity was assessed using the ALSFRS-R (Citation10) and we listed the number of regions that were affected (bulbar, upper- and lower extremities) (). None of the included patients had major comorbidity, a psychiatric disorder or another neurological disorder. Spouses, family members or caregivers were invited to fill out caregiver behavioral interviews. The study was approved by the Medical Ethical Committee of UMC Utrecht and written informed consent was obtained from all participants.
Table 1 Demographic and clinical characteristics.
Full neuropsychological evaluation
We administered a standardized neuropsychological test battery. The following cognitive domains were assessed with the subsequent tests: (1) Executive functions: Trail Making Test (TMTFootnote1) (Citation11), Rule shift cards (part of the Behavioral Assessment of the Dysexecutive Syndrome, BADS) (Citation12), Brixton Spatial Anticipation Test (Citation13); Digit span (Citation14); (2) Social cognition: reading the mind in the eyes test (Citation15); Hinting task (Citation16); (3) Verbal fluency: a phonemic verbal fluency test (3 letters, K,O,M); (4) Language: a short version of the Boston naming test (Citation17); (5) Memory: Rey’s Auditory Verbal Learning Test (RAVLT) (Citation18), the Corsi block tapping task (Citation19) and the Visual Association Test (VAT) (Citation20); (6) Visuospatial functions: Judgement of Line Orientation (JLO) (Citation21). An overview of the neuropsychological instruments including the most relevant associated cognitive domains and processes per test, with references to the applied normative values, is given in .
Table 2 Overview neuropsychological test battery.
Additional cognitive and behavioral screening instruments
The Dutch version of the ECAS (Citation4,Citation32), FAB (Citation5) and ALS-FTD-Q (Citation6) were administered on the same day as the NPE, or less than one month before or after the NPE. The ECAS is a brief multi-domain screening tool that assesses executive functions (including social cognition), language, fluency, memory and visuospatial functions. In addition to cognitive assessment, the ECAS contains a behavioral interview based on the five behavioral domains affected in bvFTD (Citation4,Citation32,Citation33). The FAB is a short screening battery sensitive to frontal lobe dysfunction (Citation5).
The ALS-FTD-Q is another caregiver interview that contains 25 items assessing frequently observed behavioral symptoms in patients with ALS-FTD. The test is a paper questionnaire consisting of multiple-choice questions. Scores on the ALS-FTD-Q ≤ 21 are considered as normal, scores ≥ 22 indicate mild behavioral changes and scores ≥ 29 indicate severe behavioral changes (Citation6).
Classification of patients
Patients were classified according to the revised consensus criteria for the diagnosis of frontotemporal dysfunction in ALS by Strong et al. (Citation34) in the following categories: PLS with cognitive impairment (CI), PLS with behavioral impairment (BI), PLS with cognitive and behavioral impairment (CBI), PLS with behavioral variant FTD (bvFTD) and PLS with dementia (D). In addition to the Strong criteria, we also used the classification of “amnestic mild cognitive impairment (aMCI)” for patients with memory deficits that existed in isolation and that did not cause problems in daily life (Citation7,Citation35).
Analyses
Test results are reported as raw test scores, medians and ranges are reported for the raw test scores and corresponding percentiles, and the percentage of patients performing at or below the 5th percentile. Normative data were acquired from a variety of sources. Normative data was available only in the form of a cutoff value for normality for the FAB (<12) and the delayed recognition task of the RAVLT (<27). Regarding the ECAS, we used recent normative values corrected for age, gender and education (Citation32). Spearman correlations were used to test: (1) the relationship between the number of abnormal cognitive domains and disease duration, and (2) the number of abnormal cognitive domains and disease severity (as assessed by the ALSFRS-R). The same procedure was repeated with the number of abnormal behavioral domains according to the ECAS behavioral interview. Statistical analyses were carried out using R (version 3.5.1).
Results
Of a total of 37 PLS patients that were approached for this study, 7 were excluded. Reasons for exclusion were severe dysarthria (2 patients), change of diagnosis to another unrelated disease (1 patient) and the likely presence of an anxiety disorder or depression, based on a score of ≥ 11 on the anxiety and/or depression subscale of the Hospital Anxiety and Depression Scale (HADS)(4 patients) (Citation36). Of the 30 included patients 21 had been included in our aforementioned study using cognitive screening instruments (Citation7).
Not all patients were capable of completing the full test battery for the following reasons; fatigue (7%), impairment of the upper extremities (17%) and severe bulbar impairment (10%). In several cases, there was no spouse/caregiver we could interview and, therefore, we could not perform a behavioral assessment (20%). An overview of test results is provided in and .
Table 3 Neuropsychological test results.
Table 4 Neuropsychological profiles of individual PLS patients based on the NPE and the ECAS behavioral interview.
Overall, only 7 out of 30 patients scored completely within the normal range on all tests. This means that in 77% of PLS patients we found an abnormal performance on: (1) at least one cognitive domain and/or (2) at least two behavioral domains on the ECAS behavioral interview and/or (3) the ALS-FTD-Q (score of ≥22). Impairment was found in the following cognitive domains: social cognition (affective theory of mind: 44%, cognitive theory of mind: 12%), fluency (27%), executive functions (cognitive flexibility: 20%; working memory: 10%) and memory (short and long-term memory: 20% and 24% of patients, respectively). Deficits in language and visuospatial functions appeared to be relatively rare (abnormal in 7% and 3% respectively).
The ECAS behavioral interview showed that loss of sympathy was the most frequent behavioral sign (20%) followed by apathy (13%), disinhibition (10%), hyperorality (10%) and perseverative or stereotyped behavior (10%). The ALS-FTD-Q indicated mild behavioral changes in 3 patients (13%) and severe behavioral changes in 4 patients (17%).
Patients were subsequently categorized according to the revised Strong consensus criteria (). In this cohort 20% of patients fulfilled the criteria for cognitive impairment (CI), 10% had behavioral impairment (BI), cognitive and behavioral impairment (CBI) was seen in 3.3% and 16.7% of patients met the formal criteria for bvFTD.
Figure 1 Classification of patients according to the revised Strong consensus criteria. *The proportion of the total of patients (n = 30) included in the study categorized according to the revised consensus criteria for the diagnosis of frontotemporal dysfunction in amyotrophic lateral sclerosis (ALS) by Strong, et al.[Citation34]. bvFTD: behavioral variant frontotemporal dementia; BI: behavioral impairment; CBI: cognitive and behavioral impairment; CI: cognitive impairment; D: ALS with dementia, not typical of FTD; *aMCI: amnestic mild cognitive impairment, not part of the revised Strong consensus criteria.
![Figure 1 Classification of patients according to the revised Strong consensus criteria. *The proportion of the total of patients (n = 30) included in the study categorized according to the revised consensus criteria for the diagnosis of frontotemporal dysfunction in amyotrophic lateral sclerosis (ALS) by Strong, et al.[Citation34]. bvFTD: behavioral variant frontotemporal dementia; BI: behavioral impairment; CBI: cognitive and behavioral impairment; CI: cognitive impairment; D: ALS with dementia, not typical of FTD; *aMCI: amnestic mild cognitive impairment, not part of the revised Strong consensus criteria.](/cms/asset/c2ab08ef-8db1-4eff-a1e5-29248fa67038/iafd_a_1620284_f0001_c.jpg)
In 10% of patients we found mild memory impairment that did not interfere with daily life and without deficits in other cognitive domains. These patients were considered to have amnestic mild cognitive impairment (aMCI). We classified 2 patients with dementia (6.6%), not typical of FTD (for instance, impairment in memory and social cognition in combination with stereotyped behavior). There were three patients (10%) with varying deficits of visuospatial functions, social cognition and language that we were unable to designate to any specific classification (unclassified).
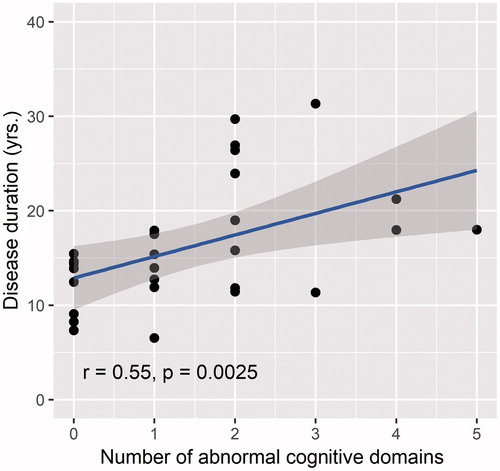
A significant correlation was observed between disease duration and the number of affected cognitive domains (r = 0.55, p = 0.0025) (). We did not observe significant correlations with disease duration and the number of abnormal behavioral domains. The presence of cognitive and/or behavioral changes was not correlated with disease severity.
Figure 2 Correlation between disease duration and the number of abnormal cognitive domains. The number of abnormal cognitive domains (performance ≤5th percentile) varied between 0–5, with a theoretical maximum of 6: executive functions, social cognition, fluency, language, memory and visuospatial functions.
Discussion
We previously reported a case series of PLS with co-morbid FTD and reviewed the literature on cognitive and behavioral changes in PLS, which suggests that changes within the FTD-spectrum are a part of the clinical phenotype of PLS (Citation3). In a follow-up study we screened a cohort of 75 PLS patients for changes within the FTD-spectrum and indeed found deficits in a considerable number of patients (10% bvFTD, 33% with BI, CI or CBI) (Citation7). These findings are in contrast with the latest revision of the El Escorial criteria that consider PLS a restricted phenotype of ALS, meaning that disease involvement is limited to upper motor neurons (Citation2). We therefore propose that PLS is not a restricted phenotype, but rather a disorder within the FTD-MND continuum (Citation37). In this current study we aimed to provide an in-depth characterization of the cognitive and behavioral profile of PLS.
A limitation of most previous studies, including our own work, is that often only simple screening instruments were used and that, therefore, cognitive deficits may have been missed. Similarly, only 5 studies assessed behavior and, therefore, behavioral changes may have been underestimated as well (Citation3). Indeed, the findings from this study show that FTD-spectrum changes are under-recognized in PLS and that in fact the frequency of these changes is very high. Only 7 out of 30 patients (23%) scored within normal range on all of the tests in our battery. We were able to classify 57% of PLS patients within one of the diagnostic categories of the revised criteria for ALS-frontotemporal spectrum disorder. In an additional 20% of cases we also identified deficits, but which were not characteristic of FTD. Although the frequency of patients with impairment in at least one domain or more is high (77%), it is important to note that it has been reported that impairment on a single domain has also been observed in 13–20% of the general population (Citation38).
It is important to note, that this present study is cross-sectional and that many patients developed cognitive and/or behavioral changes over the course of their disease (frequently many years after onset). Therefore, it seems highly plausible that the patients without cognitive and/or behavioral changes in this study may develop these as their disease progresses.
The cognitive profile of PLS we identified in this study shows deficits in social cognition, fluency, executive functions and memory. This profile is in line with that of bvFTD (Citation39,Citation40), more than the language variants of FTD (primary progressive aphasias). This is also reinforced by the observation of frequent behavioral changes in our cohort and the fact that language deficits were relatively rare (normal in 93% of patients). The diagnostic criteria for bvFTD require that memory and visuospatial functions are (relatively) spared (Citation33). It is however important to note, that in bvFTD it is episodic memory that is spared, but not necessarily other forms of memory (Citation39). Therefore the finding of memory deficits as part of the neuropsychological profile of PLS is in-keeping with bvFTD and in fact the cognitive profile is also very similar to that of ALS (Citation41).
Perhaps, the most striking observation is the high percentage of patients with deficits in social cognition. In this study social cognition was assessed using two tests for ToM, one assessing cognitive ToM and the other affective ToM (). We found a higher frequency of deficits in affective ToM (44%) versus cognitive ToM (12%), indicating that PLS patients have greater difficulty with describing the emotional/mental state of a person based on the eyes, referred to as mentalising.
This is an interesting observation as it has been argued that deficits in social cognition are highly correlated with executive dysfunction. The idea being that one first needs to recognize the emotions of others and then relies on executive functions to correctly interpret what this means for the subsequent beliefs, thought and intentions of others (Citation40). Others have, however, also suggested that changes in social cognition precede and outweigh executive dysfunction. In several case studies it has been reported that cognitive deficits on a classical NPE were subtle, while severely impaired performance was observed on ToM tests (Citation42,Citation43). We found deficits in social cognition in 12 patients, of which 7 did not have executive dysfunctions. Similarly, 5 out of 10 patients with executive dysfunction did not have deficits in social cognitions. We base this concept of dissociation between executive dysfunction and social cognition on the observation that these patients scored normally on the tasks for social cognition (Hinting task and Reading the mind in the eyes), but poorly on tasks for executive function (including those that are independent of motor slowing, such as the difference between part A and B of the TMT and the verbal fluency test) and vice versa (see Supplementary Table 1).
Therefore, our findings also suggest that deficits in social cognition are not dependent on executive dysfunction and indeed may be present at an earlier stage.
Interestingly, we previously reported a case of PLS, in which cognitive decline had been very rapid (with dementia developing over the course of a few months) (Citation3), while the motor deficits had remained relatively stable. In this study, we also seem to observe a correlation with disease duration and the development of cognitive impairment, but not with degree of disability. Although caution is warranted given the relatively small sample size of this study, it raises the hypothesis that cognitive and motor decline are independent of each other.
Considering the high frequency of FTD-spectrum deficits in PLS and the fact that these appear to develop as disease progresses, we would strongly recommend that a yearly cognitive and behavioral screening should be part of the standard clinical follow-up. Recognizing cognitive and behavioral changes is important as it is associated with noncompliance with treatment recommendations and increased caregiver burden (Citation44,Citation45). Additionally, these changes may affect capacity for medico-legal decision making and, therefore, power of attorney should be discussed early with patients (Citation46).
This study has several limitations; compared to previous cohorts, patients in our study had more disability (lower ALSFRS-R scores and more affected regions) and had longer disease duration. Furthermore, patients in our cohort were on average highly educated and may, therefore not be representative of the PLS patient population as a whole (although age and education adjusted cutoff values were applied). Another limitation of the present study is that certain cognitive functions were more extensively assessed than others (i.e. executive functions versus language and visuospatial functions). It is, therefore, possible that we underestimated the frequency of impairment for certain domains, language in particular. This also applies to subdomains; specifically we used one test to assess cognitive ToM (Hinting task) and also one to assess affective ToM (Reading the mind in the eyes); both are not necessarily comparable (range of possible scores, ceiling effect), which could have resulted in an overestimation of the gap between the percentages of impairment of cognitive and affective ToM.
In addition, reduced motor speed could have influenced test results, despite our selection of neuropsychological tests that were independent or relatively independent of motor speed. Lastly, behavioral interviews (ECAS and ALS-FTD-Q) were missing in 5 of 30 patients (17%), possibly leading to an underestimation of the number of patients with behavioral impairment.
Conclusions
In this study we performed an in-depth characterization of the cognitive and behavioral profile of PLS. We show that cognitive and behavioral changes within the FTD-spectrum occur in the majority of patients (57%) and that the cognitive profile is highly similar to that of bvFTD. We therefore conclude that PLS is not a restricted phenotype of ALS (a disease in which only upper motor neurons are involved), but rather a disorder within the FTD-MND spectrum. Clinicians should regularly assess cognition and behavior given the frequency of these types of changes and consequences it has for management, decision-making and care-giver burden. This assessment should be sensitive to the neuropsychological profile of PLS (social cognition (affective ToM in particular), fluency, executive functions and memory) and behavioral changes.
Declaration of interest
MAvE has consulted for Biogen and has received travel grants from Shire (formerly Baxalta). LHvdB reports grants from Netherlands ALS Foundation, grants from Prinses Beatrix Spierfonds, grants from Netherlands Organization for Health Research and Development (Vici scheme), grants from European Community’s Health Seventh Framework Program (FP7/2007-2013) (grant agreement no. 259867), during the conduct of the study; personal fees from Baxter for Scientific Advisory Board and Travel Grant, and personal fees from Scientific Advisory Board Biogen Idec, outside the submitted work. The other authors declare that they have no conflict of interest.
Supplementary_Table_1a.docx
Download MS Word (14.7 KB)Additional information
Funding
Notes
1 Regarding the TMT, we only used the TMT A-B score, since this score is relatively independent of motor speed.
Related Research Data
References
- Ravits J, Appel S, Baloh RH, Barohn R, Rix Brooks B, Elman L. Deciphering amyotrophic lateral sclerosis: what phenotype, neuropathology and genetics are telling us about pathogenesis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:5–18.
- Ludolph A, Drory V, Hardiman O, Nakano I, Ravits J, Robberecht W, et al. A revision of the El Escorial criteria - 2015. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:291–2.
- de Vries BS, Rustemeijer LMM, van der Kooi AJ, Raaphorst J, Schröder CD, Nijboer TCW, et al. A case series of PLS patients with frontotemporal dementia and overview of the literature. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:534–48.
- Abrahams S, Newton J, Niven E, Foley J, Bak TH. Screening for cognition and behaviour changes in ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:9–14.
- Dubois B, Slachevsky A, Litvan I, Pillon B. The FAB: a frontal assessment battery at bedside. Neurology. 2000;55:1621–6.
- Raaphorst J, Beeldman E, Schmand B, Berkhout J, Linssen WHJP, van den Berg LH, et al. The ALS-FTD-Q: a new screening tool for behavioral disturbances in ALS. Neurology. 2012;79:1377–83.
- de Vries BS, Rustemeijer LMM, Bakker LA, Schröder CD, Veldink JH, van den Berg LH, et al. Cognitive and behavioural changes in PLS and PMA: challenging the concept of restricted phenotypes. J Neurol Neurosurg Psychiatry. 2019;90:141–7.
- Gordon PH, Cheng B, Katz IB, Pinto M, Hays AP, Mitsumoto H, et al. The natural history of primary lateral sclerosis. Neurology. 2006;66:647–53.
- Pringle CE, Hudson AJ, Munoz DG, Kiernan JA, Brown WF, Ebers GC, et al. Primary lateral sclerosis. Clinical features, neuropathology and diagnostic criteria. Brain. 1992;115:495–520.
- Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci. 1999;169:13–21.
- Battery AIT. Manual of directions and scoring. Washington, DC: War Department, Adjutant General’s Office; 1944.
- Wilson BA, Alderman N, Burgess PW, et al. Behavioural assessment of the dysexecutive syndrome. Bury St. Edmunds, UK: Thames Valley Test Company; 1996.
- Burgess PW, Shallice T. The Hayling and Brixton tests. Bury St. Edmunds, UK: Thames Valley Test Company; 1997.
- Wechsler, D. (2008). Wechsler Intelligence Scale–Fourth Edition (WAIS-IV): Technical and interpretive manual. San Antonio, TX: Pearson.
- Baron-Cohen S, Jolliffe T, Mortimore C, Robertson M. Another advanced test of theory of mind: evidence from very high functioning adults with autism or asperger syndrome. J Child Psychol Psychiatry. 1997;38:813–22.
- Corcoran R, Mercer G, Frith CD. Schizophrenia, symptomatology and social inference: investigating ‘theory of mind’ in people with schizophrenia. Schizophr Res. 1995;17:5–13.
- Kaplan EF, Goodglass H, Weintraub S. The Boston Naming Test (exp. ed.). Philadelphia: Lea Febiger; 1978.
- Lezak MD. Neuropsychological assessment. New York, NY: Oxford University Press; 1976.
- Carlesimo GA, Fadda L, Lorusso S, Caltagirone C. Verbal and spatial memory spans in Alzheimer’s and multi-infarct dementia. Acta Neurol Scand. 1994;89:132–8.
- Lindeboom J, Schmand B, Tulner L, et al. Visual association test to detect early dementia of the Alzheimer type. J Neurol Neurosurg Psychiatry. 2002;73:126–33.
- Benton AL, Varney NR, Hamsher KD. Visuospatial judgment. A clinical test. Arch Neurol. 1978;35:364–7.
- Schmand B, Houx P, Koning I. Normen van psychologische tests voor gebruik in de klinische neuropsychologie. Dutch Assoc Psychol. 2012; Retrieved from www.psynip.nl
- Wilson BA, Evans JJ, Emslie H, Alderman N, Burgess P. The development of an ecologically valid test for assessing patients with a dysexecutive syndrome. Neuropsychol Rehabil. 1998;8:213–28.
- Wechsler D. WAIS-IV-NL: Wechsler Adult Intelligence Scale - Fourth Edition: Nederlandstalige bewerking: Technische handleiding en Afname en scoringshandleiding. Amsterdam: Pearson Assessment and Information B.V; 2012.
- van den Berg E, Nys GMS, Brands AMA, Ruis C, van Zandvoort MJE, Kessels RPC, et al. The Brixton spatial anticipation test as a test for executive function: validity in patients groups and norms for older adults. J Int Neuropsychol Soc. 2009;15:695–703.
- Gorissen-de Eenige M. Lees-de-ogen-test (reading the mind in the eyes test) bij volwassenen met autisme. Psychopraxis. 2007;5:204–8.
- Schmand B, Groenink SC, den Dungen M. Letterfluency: psychometrische eigenschappen en Nederlandse normen. GEEG. 2008;39:64–74.
- Loon-Vervoorn WA, van, Stumpel HJ. De Boston Benoemingstaak: Een test voor woordvinding bij afasie. Normering voor Nederland. Utrecht: Universiteit Utrecht, Vakgroep Psychonomie; 1994.
- Kessels RP, van Zandvoort MJ, Postma A, Kappelle LJ, de Haan EH. The Corsi block-tapping task: standardization and normative data. Appl Neuropsychol. 2000;7:252–8.
- Benton AL, Hamsher K, Varney NR, et al. Contributions to neuropsychological assessment: a clinical manual. New York: Oxford; 1983.
- Byom LJ, Mutlu B. Theory of mind: mechanisms, methods, and new directions. Front Hum Neurosci. 2013;7:413.
- Bakker LA, Schröder CD, Spreij LA, et al. Derivation of norms for the Dutch version of the Edinburgh cognitive and behavioral ALS screen. Amyotroph Lateral Scler Front Degener. 2018;20(1–9):19–27.
- Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456–77.
- Strong MJ, Abrahams S, Goldstein LH, Woolley S, Mclaughlin P, Snowden J, et al. Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Front Degener. 2017;18:153–74.
- Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–94.
- Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand. 1983;67:361–70.
- Burrell JR, Halliday GM, Kril JJ, Ittner LM, Götz J, Kiernan MC, et al. The frontotemporal dementia-motor neuron disease continuum. Lancet. 2016;388:919–31.
- Crawford JR, Garthwaite PH, Gault CB. Estimating the percentage of the population with abnormally low scores (or abnormally large score differences) on standardized neuropsychological test batteries: a generic method with applications. Neuropsychology. 2007;21:419–30.
- Beeldman E, Raaphorst J, Klein Twennaar M, Govaarts R, Pijnenburg YAL, de Haan RJ, et al. The cognitive profile of behavioural variant FTD and its similarities with ALS: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2018;89:995–1002.
- Harciarek M, Cosentino S. Language, executive function and social cognition in the diagnosis of frontotemporal dementia syndromes. Int Rev Psychiatry. 2013;25:178–96.
- Beeldman E, Raaphorst J, Klein Twennaar M, de Visser M, Schmand BA, de Haan RJ, et al. The cognitive profile of ALS: a systematic review and meta-analysis update. J Neurol Neurosurg Psychiatry. 2016;87:611–9.
- Lough S, Gregory C, Hodges JR. Dissociation of social cognition and executive function in frontal variant frontotemporal dementia. Neurocase. 2001;7:123–30.
- Lough S, Hodges JR. Measuring and modifying abnormal social cognition in frontal variant frontotemporal dementia. J Psychosom Res. 2002;53:639–46.
- Hu WT, Shelnutt M, Wilson A, Yarab N, Kelly C, Grossman M, et al. Behavior matters—cognitive predictors of survival in amyotrophic lateral sclerosis. PLoS One. 2013;8:e57584.
- Woolley SC, York MK, Moore DH, Strutt AM, Murphy J, Schulz PE, et al. Detecting frontotemporal dysfunction in ALS: utility of the ALS Cognitive Behavioral Screen (ALS-CBS). Amyotroph Lateral Scler. 2010;11:303–11.
- Khin Khin E, Minor D, Holloway A, Pelleg A. Decisional capacity in amyotrophic lateral sclerosis. J Am Acad Psychiatry Law. 2015;43:210–7.