
Abstract
Variants of the oxygen free radical scavenging enzyme superoxide dismutase-1 (SOD1) are associated with the neurodegenerative disease amyotrophic lateral sclerosis (ALS). These variants occur in roughly 20% of familial ALS cases, and 1% of sporadic ALS cases. Here, we identified a novel SOD1 variant in a patient in their 50s who presented with movement deficiencies and neuropsychiatric features. The variant was heterozygous and resulted in the isoleucine at position 149 being substituted with a serine (I149S). In silico analysis predicted the variant to be destabilizing to the SOD1 protein structure. Expression of the SOD1I149S variant with a C-terminal EGFP tag in neuronal-like NSC-34 cells resulted in extensive inclusion formation and reduced cell viability. Immunoblotting revealed that the intramolecular disulphide between Cys57 and Cys146 was fully reduced for SOD1I149S. Furthermore, SOD1I149S was highly susceptible to proteolytic digestion, suggesting a large degree of instability to the protein fold. Finally, fluorescence correlation spectroscopy and native-PAGE of cell lysates showed that SOD1I149S was monomeric in solution in comparison to the dimeric SOD1WT. This experimental data was obtained within 3 months and resulted in the rapid re-classification of the variant from a variant of unknown significance (VUS) to a clinically actionable likely pathogenic variant.
Keywords:
Introduction
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive neuromuscular disease in which motor neurons within the upper and lower spinal cord degenerate (Citation1). The loss of motor neurons leads to weakness, spasticity, and eventually paralysis, often resulting in patient death roughly 2.5 years after diagnosis (Citation2). The majority of ALS cases are sporadic (90%), however, some are associated with a familial history of the disease (FALS; 10% of cases). Of the FALS cases, around 20% are associated with mutations in the cytosolic oxygen free radical scavenger protein superoxide dismutase-1 (SOD1) (Citation3). There are now over 220 SOD1 mutations listed to be ALS-associated in the ALS online database, with the majority of these mutations being single site substitutions spread throughout the protein sequence (Citation4). The major hypothesis for how mutations in SOD1 cause ALS is that mutations destabilize SOD1 folding stability, resulting in misfolding and aberrant protein aggregation – reviewed in (Citation5,Citation6). Indeed, there appears to be a strong correlation between predictions of folding stability/aggregation potential and patient disease duration, where more unstable/aggregation-prone variants of SOD1 often result in more rapid disease progression (Citation7).
Some SOD1 variants exist in populations without causing ALS whilst others are known to invariably cause ALS (Citation8). A difficulty in the clinical landscape of SOD1-associated ALS has been acquiring evidence to accurately classify rare/novel variants of SOD1 in ALS patients as pathogenic on the basis of clinical evidence alone (Citation9). Therefore, molecular and biochemical analyses need to be utilized to aid in the classification of rare/novel variants to enhance outcomes for patients. Rapid and accurate classification allows patients to be more clearly diagnosed and more easily access appropriate care (Citation10). Additionally, the most promising current ALS treatment is an antisense oligonucleotide targeted against SOD1 (tofersen) (Citation11), meaning that if a patient can be identified they may receive treatment quicker. Furthermore, some clinical trials are aiming to treat pre-symptomatic patients with tofersen (NCT04856982), in which knowledge about more SOD1 variants is essential in expanding trial coverage and potentially affording greater efficacy. Therefore, methods to quickly and accurately classify SOD1 variants are of high importance to facilitating understanding and treatment of ALS.
Here, a patient in their 50s presented with clinically manifest neurological and movement deficiencies. Specifically, the patient suffered from a chronic gradually progressive form of a neurodegenerative disorder characterized by choreiform movement, anterior horn cell changes, frontal lobe cognitive changes, neuropsychiatric features, right sided weakness, and unsteady gait. Genetic sequencing identified a novel SOD1 variant that resulted in the isoleucine at position 149 mutating to serine (I149S) in the primary sequence. Molecular and biochemical analysis of SOD1I149S showed that the variant readily formed inclusions in NSC-34 cells, similar to other known SOD1-FALS variants. NSC-34 cells expressing SOD1I149S had significantly reduced viability compared to cells expressing wild-type SOD1 (SOD1WT) Furthermore, our biochemical analysis showed that SOD1I149S did not form the intramolecular disulphide, did not form dimers, and was substantially destabilized in comparison to SOD1WT. Collectively, these data were used to reclassify the variant from uncertain significance to likely pathogenic in under 3 months from identification of the variant via sequencing.
Results
SOD1I149S is predicted to be destabilizing and decrease aggregation
SOD1 monomers adopt a well-structured and highly stable eight-stranded Greek key conformation when fully mature (Citation5,Citation6). A crystal structure of SOD1 shows that I149 is oriented so that the sidechain is positioned within the hydrophobic core of the protein (), suggesting that its mutation to a polar residue could potentially disturb residue I149’s contribution to the formation of the SOD1 hydrophobic core (Citation12). Additionally, SOD1 contains multiple amyloidogenic regions within its sequence, one of which is localized to the far C-terminus, incorporating 147GVIGIAQ153 (Citation13). The input of the sequences for SOD1I149S and SOD1WT into the amyloid prediction tool Aggrescan 2D (Citation14) showed that the I149S substitution is predicted to reduce the amyloidogenicity of the C-terminus of SOD1 (), but not completely.
Figure 1. In silico analysis of the effect of mutation at position I149S of SOD1 predicts lower aggregation propensity but greater instability. (A) Side and bottom views of the SOD1 monomer structure showing residue I149 as spheres (blue) oriented into the core of the beta-barrel. (B) Aggrescan 2D measurement of wild-type (WT; black) and I149S (blue) on the aggregation propensity of SOD1. (C) MAESTRO predicted folding stability changes of the mutants at position I149 in SOD1 and A4V, showing that mutation at I149 is predicted to be destabilizing.
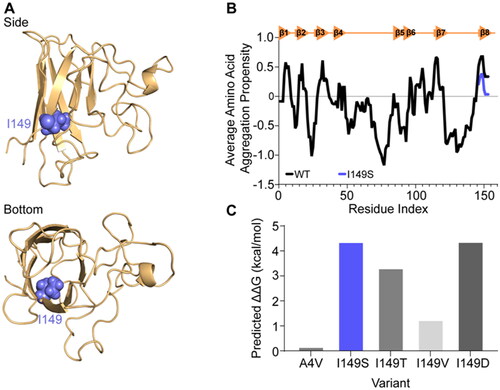
Further modeling was performed using the MAESTRO web server (Citation15) for prediction of the effect of the SOD1I149S variant on the stability of the protein. We also incorporated several variants into the predictions including known ALS-causing variants (SOD1I149T, SOD1I149V, and SOD1A4V) and a synthetic variant, SOD1I149D. MAESTRO predicted that SOD1I149S was a highly destabilizing mutant, resulting in a predicted ΔΔG value of 4.35 kcal/mol (). In comparison, SOD1A4V is predicted by MAESTRO to destabilize the protein by 0.15 kcal/mol, suggesting that SOD1I149S may be potentially more pathogenic (). Other disease-causing variants localized to I149 are SOD1I149T and SOD1I149V. We tested these variants in MAESTRO as well, finding that each were predicted to be destabilizing, with SOD1I149T decreasing predicted stability by 3.30 kcal/mol and SOD1I149V decreasing stability by 1.23 kcal/mo (). We also designed a mutation that would introduce charge into the SOD1 beta barrel core, I149D, which was predicted to destabilize the SOD1 fold with a ΔΔG value of 4.36 kcal/mol.
SOD1I149S forms inclusions and is toxic in a motor neuron-like cell model of SOD1-FALS
Previous reports have shown that when SOD1 mutants are overexpressed in established cell lines, they can form insoluble inclusions similar to those found in patients postmortem (Citation16). Indeed, this cellular model is used routinely to investigate mutations and the effect of small molecules on SOD1 inclusion formation and toxicity (Citation17). In order to determine if the SOD1I149S variant led to inclusion formation, we transiently expressed it in NSC-34 cells with a C-terminal EGFP tag alongside other variants SOD1WT, SOD1A4V, SOD1I149D, SOD1I149T, and SOD1I149V. All of the variants are known FALS mutants except for SOD1I149D which is a synthetic variant designed to maximally destabilize the I149 site. Fluorescence microscopy and automated image analysis were used to determine the percentage of cells that contained inclusions and enumerate the number of GFP-positive cells across time to determine relative cell numbers (Citation18).
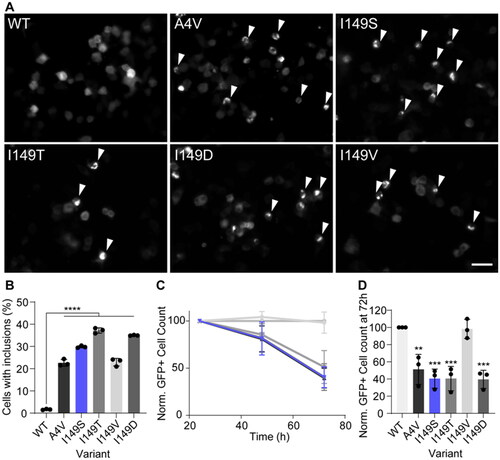
We measured inclusion formation in the top 10% highest expressing cells (determined by measuring GFP intensity per cell), as we have previously determined this population to be the most likely to form inclusions (Citation19). We observed that all variants formed significantly more inclusions than SOD1WT (p < 0.001) after 48 h post-transfection (). Expression of the novel mutant SOD1I149S resulted in approximately 30% of cells containing inclusions, which was higher than those cells expressing SOD1A4V, where approximately 23% of cells contained inclusions. The relative effect of the SOD1 variants on cell health was determined by enumerating the number of GFP-positive cells at 24 h, 48 h, and 72 h post-transfection and normalizing the values for each mutant to 24 h counts, and the SOD1WT counts (). From this, we observed that variants SOD1A4V, SOD1I149T, SOD1I149S and SOD1I149D all resulted in a loss of GFP-positive cells over time, indicating that those cell populations had their growth affected by the expression of these variants (). SOD1I149V did not differ substantially from SOD1WT at any time point, indicating it was not affecting cell growth. Statistical analysis of the GFP-positive cell counts from the 72 h time point showed that SOD1A4V, SOD1I149T, SOD1I149S and SOD1I149D were significantly lower at this point when compared to SOD1WT ().
Figure 2. SOD1I149S forms inclusions and decreases the viability of motor neuron-like cells. (A) Example images of NSC-34 cells expressing GFP-tagged SOD1 variants. Arrows point to cells containing inclusions. Scale bar = 50 µm. (B) The percentage of cells containing inclusions within the top 10% highest expressing cells, showing the I149S readily forms inclusions. (C) Enumeration of GFP + cells 24, 48, and 72 h post-transfection normalized to 24 h and wild-type counts, showing that cells expressing I149S (blue) decrease over time. (D) Quantification of the 72 h time point from panel C, showing that at 72 h, SOD1-I149S expressing cells are at 40% the number of cells expressing SOD1-WT. Error bars represent SD from at least 3 biological replicates. Significance determined by One-way ANOVA with Tukey’s post test. **** = p < 0.0001, *** = p < 0.001, and ** = p < 0.01.
SOD1I149S is highly destabilized when expressed in a cell model of SOD1-FALS
The primary consequence of mutations in SOD1 is the destabilization of the protein fold through various interconnected biophysical features of the protein (Citation5,Citation6). These features include the general folding stability of the protein, the intramolecular disulphide bond, and dimerization. All of these features can be measured biochemically to determine how a mutation may be affecting the folding state of SOD1 in cells.
To examine the consequence of the variants on SOD1 disulphide formation, we exploited the differential electrophoretic mobility of disulphide bonded SOD1 in comparison to reduced SOD1 () (Citation20). We found that both SOD1WT and SOD1I149V had similar levels of disulphide formation at approximately 40% of measured SOD1. Meanwhile, SOD1A4V showed significantly lower, but still measurable, levels of disulphide formation at approximately 10% of protein. Only around 2% of SOD1I149T was found to be disulphide bonded, indicating severe alteration to the ability of SOD1I149T to undergo or maintain this post-translational modification. Both SOD1I149S and SOD1I149D had virtually no detectable levels of disulphide bonded SOD1, further indicating a substantial effect of these mutations on this important post-translational modification. Statistical analysis showed that all variants except SOD1I149V had significantly less disulphide bonded SOD1 present (p < 0.0001).
Figure 3. SOD1I149S destabilizes the intramolecular disulphide and decreases dimer formation. (A) Immunoblotting for disulphide bonded SOD1 was carried out by running non-reducing SDS-PAGE, where faster migrating bands (SS) are disulphide bonded SOD1 and slower migrating bands (SH) are disulphide reduced SOD1. (B) Quantification of the percentage of SOD1 that was disulphide bonded showing that SOD1I149S was completely reduced. (C) Brightness histograms from fluorescence correlation spectroscopy showing that the average brightness of SOD1WT and SOD1I149V were higher than other variants, indicating these two variants are oligomeric. (D) Clear Native-PAGE of transfected cell lysates showing GFP signal where several species are observable (HMW = high molecular weight). Only SOD1WT and SOD1I149V showed any dimer signal. (E) Quantification of the percentage of each species present from clear Native-PAGE in panel (D). (F) Limited proteolysis of SOD1 variants using proteinase-K showed that the variants were differentially susceptible to proteolysis over 1 h. Error bars represent SD of the mean from 3 separate experiments. Significance determined with One-way ANOVA using Tukey’s post-hoc test (p < 0.0001 = ****).
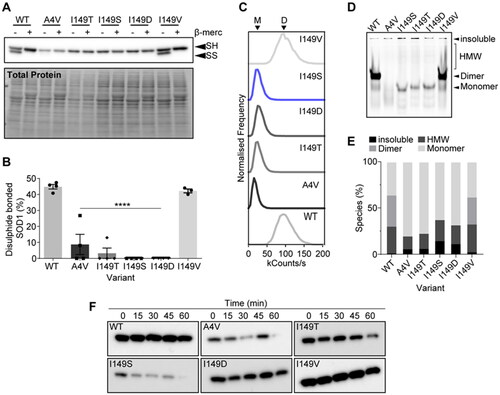
We next examined the effect of the various mutations on the ability of SOD1 to form native dimers. To do this, we utilized fluorescence correlation spectroscopy (FCS) and clear Native-PAGE. FCS is a technique that can determine the molecular brightness of molecules in solution (Citation21). In our case, a dimer of SOD1-EGFP would be expected to be brighter than a monomer. We observed that both SOD1WT and SOD1I149V had a molecular brightness of roughly 100 kcounts/s, whereas the other variants all were found to have molecular brightness readings of 25 kcounts/s (). The lower count rate for some variants indicates that the detected molecules produced less photons, suggesting fewer GFP molecules (monomers) were being detected for a single event. In conjunction, clear Native-PAGE separated SOD1-EGFP variants on their charge and hydrodynamic volume, where both SOD1WT and SOD1I149V showed an intense band that represents dimer (). Other variants did not show this band, but rather only monomer bands, high molecular weight smears and signal in the gel wells that is insoluble material too large to enter the gel matrix. Together, FCS and clear Native-PAGE indicate that soluble SOD1I149S is monomeric in cell lysates.
Finally, we examined the susceptibility of these SOD1 variants to proteolytic digestion from proteinase-K. Past research has shown that SOD1WT is resistant to proteolysis whereas ALS-associated variants are susceptible to proteolysis due to the structural destabilization induced by mutation to the protein (Citation22). Lysates from cells expressing SOD1 variants were harvested and incubated for different times prior to immunoblotting for the detection of SOD1-EGFP. We found that SOD1WT and SOD1I149V were both resistant to proteolytic digestion via proteinase-K. SOD1I149D was only partially susceptible to digestion, whereas SOD1A4V, SOD1I149T, and SOD1I149S were all susceptible to proteolytic digestion, indicating these variants are highly structurally destabilized.
Discussion
In this work we identified a novel SOD1 mutation I149S, which we compared in cell and biochemical assays to several other variants, including those occurring at the same amino acid position. I149 is located on beta-strand 8 of the SOD1 protein, which takes part in the formation of the SOD1 dimer interface and is proximal to C146, which is responsible for intramolecular disulphide bond formation (Citation5,Citation6). Consequently, we measured that the SOD1I149S variant substantially altered these two features of the SOD1 structure compared to SOD1WT. Impaired dimerization (Citation23) and the presence of reduced SOD1 (Citation24) are hallmarks of other pathological variants of SOD1. Indeed, SOD1I149S was found to be even more destabilizing than the prevalent and rapidly progressive SOD1A4V variant in our assays. Curiously, SOD1I149V behaved similarly to SOD1WT in all assays except the inclusion formation assay. Our expectation was that SOD1I149V would be less destabilizing due to the amino acid substitution maintaining a hydrophobic residue. We did not expect such extensive inclusion formation without other features of the protein being affected. Previous research has suggested that the presence of inclusions does not always correlate with disease, and our analysis of SOD1I149V supports this (Citation25). Overall, our assays were capable of capturing the diverse outcomes that occur with even just different residue substitutions at the same site in SOD1.
Using our robust and comprehensive assays, in under 3 months, we were able to effectively reclassify SOD1I149S as ‘likely pathogenic’ according to ACMG/AMP criteria (Citation9). As a result, the patient now has a clear diagnosis, an understanding on his prognosis, potential access to clinical trials, and his relatives now have the choice of predictive testing. Currently, around 50% of known SOD1 variants are listed as only having in silico predictions of their pathogenicity, which is not recommended under the ACMG/AMP criteria (considered the 3rd lowest level of evidence; PP3). Rather, the ACMG/AMP criteria place heavier weighting on the usage of well-established functional assays which form the 3rd strongest level of evidence (PS3). It has been three decades since mutations in SOD1 were the first identified genetic linkage to ALS (Citation26). Since then, over 220 SOD1 variants have been linked to ALS with varying degrees of evidence to support their pathogenicity. Roughly 7 new variants are identified every year, yet the evidence needed to accurately classify these variants according to standard ACMG/AMP criteria is lacking. Often, mutations are identified in patients but not characterized for many years, or at all, due to poor communication between clinicians and basic scientists, and a lack of effective and rapid models for variant phenotype assessment (Citation27).
Considering the most promising therapies in clinical trials for ALS are currently targeted at SOD1 (Citation11), increasing the entry of patients into these clinical trials is highly important. Another clinical trial that has recently started in Australia is the ATLAS trial, which aims to use the SOD1-targeting antisense therapy, tofersen, on pre-symptomatic SOD1 variant carrying patients (NCT04856982). Prior knowledge and accurate classification of SOD1 variants is essential if we are to begin treating SOD1-FALS in people prior to symptom onset. Our methods used here are cheap and effective for this purpose. A more effective approach with predictive capacity would be to carry out a multiplexed analysis of variant effect (MAVE) study on SOD1 to determine the effect of all possible variants (Citation28). A MAVE assay of SOD1 variants should be designed to measure the multiple biophysical parameters important for pathogenicity, such as folding stability, maturation status, and aggregation. Such a dataset would allow for the classification of all existing variants of unknown significance (VUS) and future identified variants to improve patient knowledge and outcomes far quicker than reactive classification as we have done here.
We propose that clinicians and basic researchers should more readily work together to solve patient-specific problems with diagnosis and treatment. Specifically, once a novel variant is identified, clinicians should engage with biomedical scientists to notify them of the variant. Following this, biomedical scientists should assay the variant in comparison to multiple known pathogenic variants (e.g. A4V) and benign SOD1 (e.g. WT) in cell lines, as per the Clinical Genome Resource Sequence Variant Interpretation Working Group guidelines (Citation29). Experiments that determine dimer affinity, disulphide status, metalation state, folding stability, and aggregation potential should all be attempted if possible, as the state of these parameters are often variant-specific. This will create a profile for the variant so it can be potentially classified similar to how we did here. Of course, researchers often use different models (plasmids, cell lines) which may generate different results, but we believe that as long as good positive and negative controls are present, effective comparisons can be made (Citation29). Doing this will enhance knowledge surrounding SOD1-associated ALS and provide clearer diagnoses for those patients with a less well-defined presentation of ALS.
Authors contributions
Conceptualization: LM, BT, AH; Methodology: LM, VS, MB, BT, AH; Validation: LM, VS, MB, BT, AH; Formal analysis: LM, VS, MB, BT, AH; Investigation: LM, VS, MB, BT, AH; Resources: LM, BT, AH; Data curation: LM, VS, MB, BT, AH; Writing - original draft: LM; Writing – review & editing: LM, VS, MB, BT, AH; Visualization: LM; Supervision: LM; Project administration: LM, BT, AH; Funding acquisition: LM.
Supplemental Material
Download MS Word (27.4 KB)Acknowledgements
LM acknowledges funding from Motor Neuron Disease Australia and FightMND. The authors thank the patient and their family for their contribution to this work. The authors would like to pay deep respects to the late Professor Justin J. Yerbury (University of Wollongong, Australia), without whom this work would not have been done and authors LM, VS, and MB owe immeasurable gratitude.
Declaration of interest
The Human Research Ethics Committee (HREC) of Royal Melbourne Hospital (RMH, Victoria Australia) waived ethical approval for this submission. Written informed consent for participation in genetic testing and the publication of molecular and clinical data was obtained from the patient.
Additional information
Funding
References
- Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17071.
- Masrori P, Van Damme P. Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol. 2020;27:1918–29.
- Taylor JP, Brown RH, Cleveland DW. Decoding ALS: from genes to mechanism. Nature 2016;539:197–206.
- Abel O, Powell JF, Andersen PM, Al-Chalabi A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum Mutat. 2012;33:1345–51.
- Wright GSA, Antonyuk SV, Hasnain SS. The biophysics of superoxide dismutase-1 and amyotrophic lateral sclerosis. Q Rev Biophys. 2019;52:e12.
- McAlary L, Plotkin SS, Yerbury JJ, Cashman NR. Prion-like propagation of protein misfolding and aggregation in amyotrophic lateral sclerosis. Front Mol Neurosci. 2019;12:262.
- Wang Q, Johnson JL, Agar NYR, Agar JN. Protein aggregation and protein instability govern familial amyotrophic lateral sclerosis patient survival. PLOS Biol. 2008;6:e170.
- Andersen PM, Restagno G, Stewart HG, Chiò A. Disease penetrance in amyotrophic lateral sclerosis associated with mutations in the SOD1 gene. Ann Neurol. 2004;55:298–9; author reply 9.
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
- Watts G, Newson AJ. Is there a duty to routinely reinterpret genomic variant classifications? J Med Ethics. 2023;49:808–14.
- Miller TM, Cudkowicz ME, Genge A, Shaw PJ, Sobue G, Bucelli RC, et al. Trial of antisense oligonucleotide tofersen for ALS. N Engl J Med. 2022;387:1099–110.
- Strange RW, Antonyuk S, Hough MA, Doucette PA, Rodriguez JA, Hart PJ, et al. The structure of holo and metal-deficient wild-type human Cu, Zn superoxide dismutase and its relevance to familial amyotrophic lateral sclerosis. J Mol Biol. 2003;328:877–91.
- Ivanova MI, Sievers SA, Guenther EL, Johnson LM, Winkler DD, Galaleldeen A, et al. Aggregation-triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. Proc Natl Acad Sci U S A. 2014;111:197–201.
- Conchillo-Solé O, de Groot NS, Avilés FX, Vendrell J, Daura X, Ventura S. AGGRESCAN: a server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinf. 2007;8:65.
- Laimer J, Hofer H, Fritz M, Wegenkittl S, Lackner P. MAESTRO–multi agent stability prediction upon point mutations. BMC Bioinformatics. 2015;16:116.
- McAlary L, Aquilina JA, Yerbury JJ. Susceptibility of mutant SOD1 to form a destabilized monomer predicts cellular aggregation and toxicity but not aggregation propensity. Front Neurosci. 2016;10:499.
- McAlary L, Shephard VK, Wright GSA, Yerbury JJ. A copper chaperone-mimetic polytherapy for SOD1-associated amyotrophic lateral sclerosis. J Biol Chem. 2022;298:101612.
- McAlary L, Shephard VK, Sher M, Rice LJ, Yerbury JJ, Cashman NR, et al. Assessment of protein inclusions in cultured cells using automated image analysis. STAR Protoc. 2022;3:101748.
- Farrawell NE, Lambert-Smith I, Mitchell K, McKenna J, McAlary L, Ciryam P, et al. SOD1A4V aggregation alters ubiquitin homeostasis in a cell model of ALS. J Cell Sci 2018;131:jcs209122.
- Furukawa Y, O'Halloran TV. Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo- and reduced form of SOD1, leading to unfolding and oxidative aggregation. J Biol Chem. 2005;280:17266–74.
- Elson EL. Fluorescence correlation spectroscopy: past, present, future. Biophys J. 2011;101:2855–70.
- Ratovitski T, Corson LB, Strain J, Wong P, Cleveland DW, Culotta VC, et al. Variation in the biochemical/biophysical properties of mutant superoxide dismutase 1 enzymes and the rate of disease progression in familial amyotrophic lateral sclerosis kindreds. Hum Mol Genet. 1999;8:1451–60.
- McAlary L, Yerbury JJ, Aquilina JA. Glutathionylation potentiates benign superoxide dismutase 1 variants to the toxic forms associated with amyotrophic lateral sclerosis. Sci Rep. 2013;3:3275.
- Keskin I, Forsgren E, Lehmann M, Andersen PM, Brännström T, Lange DJ, et al. The molecular pathogenesis of superoxide dismutase 1-linked ALS is promoted by low oxygen tension. Acta Neuropathol. 2019;138:85–101.
- Gill C, Phelan JP, Hatzipetros T, Kidd JD, Tassinari VR, Levine B, et al. SOD1-positive aggregate accumulation in the CNS predicts slower disease progression and increased longevity in a mutant SOD1 mouse model of ALS. Sci Rep. 2019;9:6724.
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993;362:59–62.
- Hoffman-Andrews L. The known unknown: the challenges of genetic variants of uncertain significance in clinical practice. J Law Biosci. 2018;4:648–57.
- Gasperini M, Starita L, Shendure J. The power of multiplexed functional analysis of genetic variants. Nat Protoc. 2016;11:1782–7.
- Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019;12:3.