
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The aim of the present investigation was to fabricate and evaluate solid lipid nanoparticles (SLNs) of asenapine maleate (AM) to improve its oral bioavailability (BA). AM-SLNs were prepared by high speed homogenization followed by ultrasonication technique. The resultant SLNs exhibited particle size, zeta potential and entrapment efficiency of 114.3 ± 3.5 nm, −12.9 ± 3.8 mV, and 84.10% ± 2.90% respectively. In vitro release study of AM-SLNs showed 9.23% ± 2.72% and 92.09% ± 3.40% release of AM in pH 1.2 medium and phosphate buffer pH 6.8, respectively, indicating higher potential of lymphatic uptake. Cell viability study using Caco-2 cell line indicated non-toxicity of the carriers and drug. The uptake of AM-SLNs across Caco-2 cell line was time and energy dependent exhibiting clathrin-claveole mediated endocytosis transport. Cellular uptake of Coumarin loaded SLNs was effectively increased as compared to the dye solution. The pharmacokinetic results in rats showed 50.19-fold improvement in BA of AM after fabrication of SLNs. Collectively, all these findings demonstrated effectiveness of SLNs to improve therapeutic efficacy of AM in the treatment of schizophrenia.
Introduction
Nanoparticulate based drug delivery system has emerged as a promising approach which can boost the bioavailability (BA) of lipophilic drugs [Citation1]. Nanocarriers offer various advantages such as protection of drug degradation in the GIT, provide controlled drug release, change internalization pathway and thereby improve drug delivery across the intestinal cell monolayers [Citation2]. Different nanocarriers such as microemulsions, liposomes, polymeric nanoparticles, and solid lipid nanoparticles might overcome the drawbacks associated with poorly water-soluble drugs.
Amongst various nanocarriers, lipid-based nanoparticles are widely employed for the effective delivery for BA improvement as lipid excipients provide advantages such as increased solubility, lymphatic targeting, and/or modulation of enterocytes-based transport. Solid lipid nanoparticles (SLNs) have become an attractive carrier for the scientist to improve pitfalls of oral delivery because of their unique properties over other colloidal carriers. SLNs consist of solid lipid (biocompatible) core with entrapped drug which provides controlled drug release and improves its therapeutic efficacy [Citation3]. Zhang et al. [Citation4] prepared and evaluated SLNs of candesartan cilexetil (CC) to overcome its low BA. The authors reported that CC loaded SLNs were uptaken by enterocytes and reached to the systemic circulation by intestinal lymphatic system. The results of pharmacokinetic study proved 12-fold improvement in oral BA after fabricating SLNs.
Asenapine maleate (AM) is a second generation (atypical) antipsychotic agent which has strong inhibitory action on 5HT2A (serotonin) and D2 (dopamine) receptor. It improves cognitive function and negative symptoms in patients suffering from schizophrenia. AM belongs to BCS class II (log p values of 4.9) having oral BA <2% because it undergoes extensive first pass metabolism [Citation5]. It is also a CYP1A2 substrate. Although AM is available as sublingual tablet having BA 35%, organoleptic properties and patient compliance are limiting parameters. It was felt that orally administered solid lipid nanoparticles would enhance BA by bypassing first pass via lymphatic uptake and CYPA inhibition. Hence, this research work was intended to increase the BA by fabrication of AM-SLNs, a lipid based formulation which can avoid the first pass metabolism via intestinal lymphatic system and can enhance its oral BA [Citation3].
We have previously reported the detailed physicochemical characterization of AM-SLNs [Citation6]. It was demonstrated that process and formulation variables had an enormous influence on SLNs particle size and drug encapsulation efficiency. This work focuses on various characterization and evaluation parameters. Furthermore, cell viability, cell uptake, absorption mechanism and permeability across Caco-2 cell monolayer were elucidated. The pharmacokinetic study and intestinal lymphatic study using cycloheximide (CHX) in rats were also conducted to prove its potential in enhancement of oral BA of AM.
Materials and methods
Materials
AM was obtained from Alembic Pharmaceuticals Limited (Gujarat, India). Glyceryl monostearate (GMS) and D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) were obtained from Loba Chemie Pvt Limited (Mumbai, India) and Antares Health Products (Jonesborough, TN, USA) respectively. Penicillin–streptomycin solution, minimum essential medium Eagle (MEM), Hank’s balanced salt solution, Trypsin-EDTA solution, fetal bovine serum (FBS) were purchased from Himedia (Mumbai, India). Transwell inserts (12-well) and well plates (6 and 96) were purchased from Nunc (Roskilde, Denmark) and Costar, Corning (Washington, USA), respectively. Poloxamer 188, MK-801, CHX, sodium azide, chlorpromazine, nystatin, coumarin-6 and 4,5-(dimethylthiazol-2-yl) 2,5-diphenyl-tetrazolium bromide (MTT) dye were purchased from Sigma Aldrich (Mumbai, India). All other reagents used were of analytical grade.
Preparation of AM loaded SLNs
Briefly, GMS was melted at 70 °C and 10 mg of AM was dissolved in it. This lipid phase was dispersed in the aqueous surfactant solution (2.5% of Poloxamer 188 and 0.02% of D-α- TPGS) at 70 °C using a homogenizer (T-25 digital Ultra-Turrax, IKA India Private Limited, Bengaluru, India). The resultant emulsion was ultrasonicated using a probe sonicator (Labsonic, Sartorius, Germany) and cooled in an ice bath [Citation6,Citation7]. The obtained AM-SLNs were lyophilized using trehalose in the ratio of 1:3 with respect to solid content [Citation8]. The drug dispersion was prepared by adding 10 mg of AM in water. 6-Coumarin loaded SLNs were prepared similarly by replacing AM with 6-coumarin during preparation for cell line studies.
Physicochemical characterization of AM-SLNs
Detailed characterization of the AM-SLNs has been described previously [Citation6]. The particle size (PS) and zeta potential of the SLNs were measured using Zetasizer (Malvern, UK). The entrapment efficiency (%EE) was calculated from the following equation.
(1)
(1)
Differential scanning calorimetry (DSC)
The DSC thermograms of bulk AM, excipients (GMS, physical mixture of AM and GMS) and lyophilized AM-SLNs were taken using Differential Scanning Calorimeter (Shimadzu DSC-60) [Citation9].
X-ray powder diffraction (XRD)
XRD studies of bulk AM, formulation components (GMS, physical mixture of AM and GMS) and lyophilized AM-SLNs were carried out by X-Ray scattering technique (Philips, PW 1710) [Citation10].
Transmission electron microscopy (TEM) of AM-SLNs
TEM image of AM-SLNs was taken by taking a drop of formulation on copper grid which was treated with phosphotungstic acid and inserted in Tecnai 20 transmission electron microscope (Philips, Holland) [Citation11].
In vitro release study using dialysis bag method
AM-SLNs and AM dispersion equivalent to 10 mg AM was placed in dialysis bag having molecular weight cut off (12,000–14,000 Da) which was securely sealed from both ends and introduced into 50 ml of gastric fluid (pH 1.2) for 2 h followed by phosphate buffer pH 6.8 [Citation12]. The entire system was kept under continuous stirring at 100 rpm at 37 °C [Citation13]. Samples were withdrawn at predetermined time intervals, filtered and analyzed using UV spectroscopy at 270 nm.
Cell line studies
Cell culture
The human epithelial colorectal adenocarcinoma cell line, Caco-2 were obtained from National Centre for Cell Science (Pune, India). The cells were grown in tissue culture flasks of 75 cm2 in Jouan IG0150 incubator (Thermo-Fischer, Waltham, MA, USA) using growth medium (MEM, FBS [20%], 100 IU/ml of penicillin and 100 μg/ml of streptomycin). Fresh nutrition to the cells was provided by replacing the medium on every alternate day. Once 90% confluency was reached, the cells were harvested using trypsin-EDTA solution (0.25%) and used for subsequent studies.
Assessment of cell viability using MTT assay
The cells were seeded (1 × 104 cells/well) in 96-well plates and incubated for 24 h [Citation12]. The cells were treated with 100 μl of medium containing SLNs at different concentrations for 24 h. The supernatants were discarded, 100 μl of MTT reagent (1 mg/ml) was added to each well and further incubated with for 4 h. After 4 h, MTT was removed and 100 μl dimethyl sulfoxide was added to each well. The absorbance was measured using Microplate Reader (680-XR, Bio-Rad Laboratories, Roanne, France) at 570 nm.
Cell uptake study
Cell uptake study of coumarin-6 loaded SLNs and coumarin-6 solution (100 μg/ml) was carried out across Caco-2 cell line using confocal microscopy and flow cytometry.
For confocal microscopic analysis, 1 × 106 cells per well were seeded in 6-well plates on cover slips and incubated for 24 h. After 24 h, the cells were incubated with 100 μl of samples (coumarin-6 solution and coumarin-6 loaded SLNs) for 1 h and 4 h. Thereafter, they were washed with phosphate buffered saline (PBS) and fixed using ethanol. Images were captured using confocal laser microscope [Citation14,Citation15].
For flow cytometry, 1 × 105 cells per well were seeded in 6-well plate and incubated for 24 h prior to study. The cells were incubated with 100 μl of coumarin-6 solution and coumarin-6 loaded SLNs for 1 and 4 h. After the incubation period, they were washed with PBS and harvested using trypsin-EDTA solution. The cells were centrifuged at 1000 rpm for 5 min. After discarding the supernatants, they were collected and fluorescence was measured in FACS (FACS Canto-II, BD Biosciences, San Jose, CA, USA) [Citation16,Citation17].
Elucidation of uptake mechanism of SLNs in Caco-2 cell monolayer
The transport mechanism across Caco-2 cell monolayer was evaluated in presence of different inhibitors. Caco-2 cells were seeded at a concentration of 1 × 105 cells/well into 6-well plate and incubated for 24 h. After 24 h, the cell monolayers were incubated with culture medium containing sodium azide (0.1% w/v), chlorpromazine (10 μg/ml) and nystatin (50 μg/ml) for 1 h respectively [Citation17,Citation18]. After 1 h, AM-SLNs and AM dispersion (10 μg/ml) were added into the inhibitor solution and co-incubated for 2 h. Thereafter, the cells were lysed using Triton X-100 [Citation18]. Drug was extracted using acetonitrile and quantified by high-performance liquid chromatography (HPLC) at 210 nm [Citation19].
Transport study across Caco-2 cell monolayer
Approximately 5000 cells per well were seeded in transwell inserts (12 mm, 0.4 mm pores) and cultured for 21 days [Citation17,Citation20]. The integrity of the Caco-2 cells was checked by measuring trans epithelial electrical resistance (TEER) using EVOM2 (World Precision Instrument, Sarasota, FL, USA). Cell monolayers having TEER more than 800 Ω•cm2 were selected to carry out transport study. For apical to basolateral transport study, 0.5 ml containing 100 μg/ml each of AM-SLNs and AM dispersion (diluted with transport medium) was added to the apical side while 1.5 ml of transport buffer was added to the basolateral side. After incubation for specified time period, aliquot of 100 μl was withdrawn and amount of drug permeated was quantified using HPLC [Citation17,Citation21]. The apparent permeability coefficient (Papp) was calculated using the following equation:
(2)
(2)
where, Papp, dQ/dt, A, C0 is the apparent permeability coefficient (cm/s), steady state flux, surface area (cm2) and initial concentration of drug used respectively.
Enhancement ratio (ER) was determined using the following formula:
(3)
(3)
Oral BA study of AM-SLNs and AM dispersion
The animal study protocol [No. MSU/IAEC/2014/1438] involving use of female Sprague–Dawley (SD) rats (180–220 g) was duly approved by the Institutional Animal Ethics Committee (IAEC) of The M. S. University of Baroda, Vadodara, India. The animals were randomly divided into two groups (n = 6) and the formulations (AM dispersion and AM-SLNs) were administered orally at a dose of 1.033 mg/kg using an oral feeding cannula. 0.2 ml of blood samples were withdrawn from retro-orbital plexus post administration and collected into microcentrifuge tube containing anticoagulant (heparin). Plasma was separated immediately by centrifugation (10,000 rpm, 10 min, 4 °C) [Citation22,Citation23]. Drug was extracted using acetonitrile, injected in to the HPLC column and analyzed at 210 nm using Acetonitrile: Phosphate buffer (65:35) as a mobile phase [Citation18]. The pharmacokinetic parameters were determined by Thermo Kinetica 5.0 software. The relative BA was determined by comparing area under curve (AUC) of AM-SLNs with AUC of AM dispersion.
Lymphatic transport study of AM-SLNs using CHX
In vivo lymphatic uptake study of formulation occurs via blockage of chylomicron production or its secretion from basolateral side of the enterocyte. Hence, comparison of pharmacokinetic data of animals pretreated with a chylomicron flow blocking substance, CHX and control animals was done to confirm lymphatic transport of AM-SLNs [Citation24].
Lymphatic uptake of AM-SLNs was evaluated using CHX (a chylomicron blocker). In this study, the rats were divided into two groups (n = 6). Group I was administered with CHX (3 mg/kg) intraperitonially and group II was administered with saline [Citation25]. One hour after the injection, both groups were orally administered with AM-SLNs at a dose of 1.033 mg/kg. At defined time intervals, blood samples were collected and were processed as described earlier.
Stability study
The stability of SLNs was carried out for 3 months at 2–8 °C and Room temperature (30 ± 2 °C/60%±5% RH). Samples were withdrawn at 0, 1, 2, and 3 months and particle size, zeta potential and drug content were determined [Citation26].
Statistical analysis
Each value was expressed as mean ± standard deviation. Statistical analysis was carried out with ANOVA test and p < .05 was considered to be statistically significant differences.
Results and discussion
Physicochemical characterization of AM-SLNs
The particle size, polydispersity index and zeta potential of AM-SLNs was found to be 114.3 ± 3.5 nm, 0.188 ± 0.010, and −12.9 ± 3.8 mV, respectively. The small particle size (below 200 nm) will provide large surface area for absorption and also promote lymphatic uptake [Citation17]. The %EE was found to be 84.10% ± 2.90% owing to high solubility of AM in GMS.
Differential scanning calorimetry (DSC)
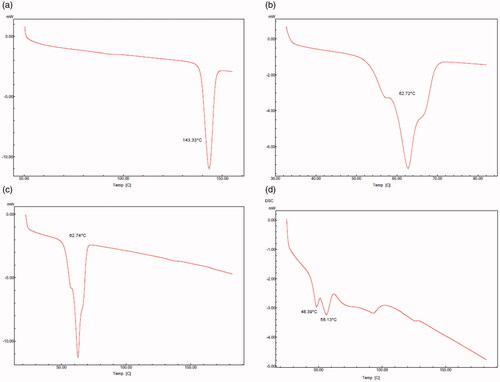
The DSC thermograms of bulk AM, excipients and lyophilized AM-SLNs are shown in . The DSC thermogram of AM showed sharp endothermic peak at 143.33 °C indicating its crystalline nature. The thermogram of GMS showed peak at 62.72 °C related to its melting point. The physical mixture of AM and GMS showed the melting endotherm at 62.74 °C corresponding to melting point of GMS. It showed absence of melting endotherm of drug indicating that AM was either completely solubilized in the GMS or amorphized. The absence of endothermic peak of AM in lyophilized AM-SLNs indicated molecular dispersion of AM in GMS or conversion from crystalline to amorphous state. Additional peak at ∼49 °C might be due to presence of surfactant (Poloxamer 188) and the peak at ∼86 °C was due to trehalose used as the cryoprotectant during lyophilization of SLN. The decrease in the peak and onset temperature (∼56 °C) of lipid can be due to reduced particle size and increased surface area which leads to reduction in melting enthalpy [Citation27].
Figure 1 DSC thermogram of (a) AM (b) GMS (c) physical mixture of AM and GMS (d) lyophilized AM-SLNs.
X-ray powder diffraction (XRD)
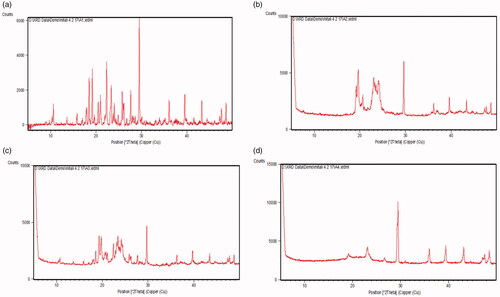
The diffractograms of bulk AM, formulation components (GMS, physical mixture of AM and GMS) and lyophilized AM-SLNs are shown in . The diffraction pattern of AM showed distinct sharp peaks at 2θ = 17.8°, 18.5°, 19.6°, 22.5°, 23.6°, 29.6°, 36.2°, 39.5°, 43.5°, and 48.7° indicating crystalline nature of AM. The diffraction pattern of GMS showed peaks at 2θ = 19.1°, 22.9°, 29.6°, 36.4°, and 39.8°. The characteristic peaks of AM could not be detected in the physical mixture of drug and lipid which indicated complete solubilization of AM in the GMS. The absence of characteristic peaks of AM in lyophilized AM-SLNs confirmed solubilization in lipid or conversion of AM into amorphous state. Moreover, this also indicated incorporation of AM within the crystal lattice of the lipid as the AM-SLNs were developed by dissolving AM in GMS which allowed the homogenous dispersion of drug in the GMS [Citation28]. It can be implied that the solubilized drug would remain encapsulated in the lipid matrix during storage.
Figure 2 XRD of (a) AM (b) GMS (c) physical mixture of AM and GMS (d) lyophilized AM-SLNs.
TEM of AM-SLNs
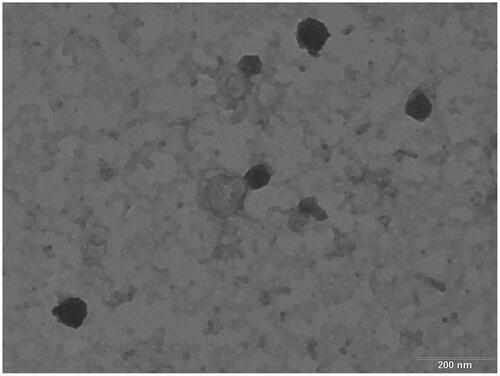
The TEM image of the AM-SLNs () typically displays spherical shape with dense lipid matrix. The particle size observed by TEM was found to be in the range of 80–120 nm which supports the results obtained with Malvern zetasizer.
Figure 3 TEM image of optimized batch of AM-SLNs.
In vitro release study using dialysis bag
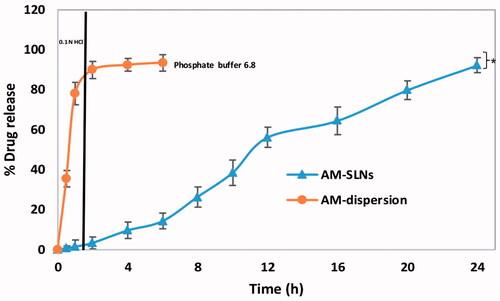
The in vitro drug release profiles of AM-SLNs and drug dispersion as a function of time are shown in . The release profile of AM dispersion indicated rapid diffusion of drug (89.92% ± 3.03%) through dialysis membrane in gastric fluid (pH 1.2) with subsequent release (93.42% ± 3.26%) in intestinal fluid (phosphate buffer pH 6.8). In contrast, drug release from AM loaded SLN was only 9.23% ± 2.72% at pH 1.2 indicating that inclusion of AM into lipid nanoparticles significantly reduced its release in pH 1.2 media followed by slow and controlled release of AM (92.09% ± 3.40%) from AM-SLNs in phosphate buffer pH 6.8 at 24 h. Thus, this release profile can be owing to the slow diffusion of AM from the SLNs matrix [Citation29]. Moreover, as previously reported, Poloxamer 188 (a swellable hydrophilic macromolecule) which forms a hydrogel barrier can also control the migration of drug from the lipid layers [Citation30,Citation31]. Among the various release models, AM-SLN fitted well to Korsmeyer Peppas (R2=0.9818) model and release mechanism was found to follow fickian (n = 0.202) diffusion. Drug release from the AM dispersion was found to be first order kinetics i.e. showed concentration dependent release.
Figure 4 In vitro drug release profile of AM-SLNs and drug dispersion (n = 3). *p < .05 compared to drug dispersion.
Cell line studies
Assessment of cell viability using MTT assay
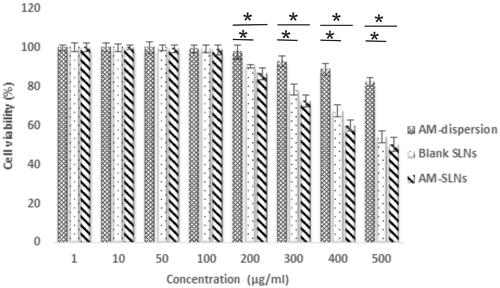
The results of cell viability assay are shown in . The cell viability was >80% for AM dispersion indicating its non-toxicity. The results showed 100% cell viabilities for blank SLNs up to 100 μg/ml concentration suggesting that the components used for SLNs were non-toxic. Moreover, greater than 70% of cell viability was observed upto 300 μg/ml while further increase in concentration significantly reduced cell viability. AM-SLNs also showed >70% cell viability upto 300 μg/ml but further increasing the concentration to 500 μg/ml drastically reduced the cell viability. Thus, no significant difference was observed in the cell viabilities of blank and drug loaded SLNs, further confirming that the drug was non-toxic. As the results demonstrated 100% cell viability upto 100 μg/ml concentration, uptake and permeation studies were conducted at that concentration.
Figure 5 Cell viability assay of the AM-SLNs in Caco-2 cell line (n = 3). *p < .05 compared to drug dispersion.
Cell uptake study
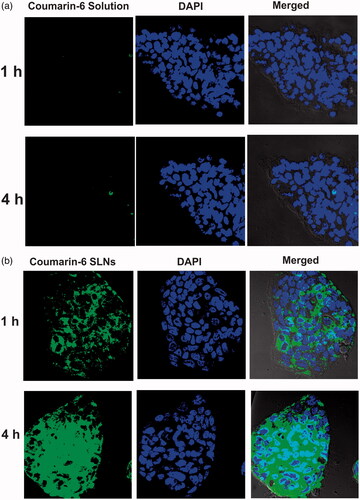
Efficient cellular uptake is the prerequisite for the therapeutic efficacy of SLNs. Confocal and flow cytometry studies were conducted to compare the cellular uptake of coumarin-6 loaded SLNs with coumarin-6 solution. Results () depicted that uptake of coumarin-6 loaded SLNs and coumarin-6 solution was time dependent. In case of dye solution, very little fluorescence was observed at the end of 4 h indicating its poor internalization into cell monolayers. However, SLNs showed more fluorescence intensity, both in the cell cytoplasm as well as in the nucleus implying that SLNs were internalized into the Caco-2 cells.
Figure 6 Cellular uptake of (a) coumarin-6 solution, (b) coumarin-6 loaded SLNs across Caco-2 cells using confocal microscopy at different time intervals.
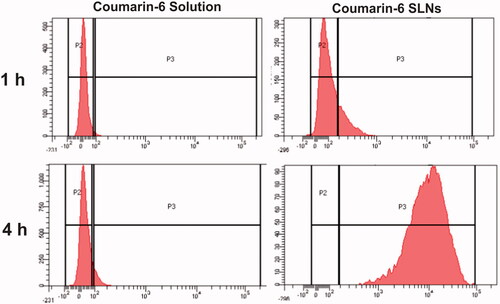
The results of flow cytometry showed that fluorescence intensity inside the cells was significantly increased (99.24 times) by coumarin-6 loaded SLNs as compared to plain dye solution (). This study also revealed that the SLNs were internalized in a time dependent manner and their uptake was effectively enhanced at the end of 4 h. This finding strongly supports the results obtained from confocal microscopy.
Figure 7 Histogram showing uptake of coumarin-6 solution, coumarin-6 loaded SLNs at different time points across Caco-2 cells.
It is reported that uptake is increased as the hydrophobic interactions of nanoparticles with Caco-2 cell membrane increased [Citation32]. Also, the enhanced permeation of SLNs can be attributed to their small particle size and presence of TPGS, a potential absorption enhancer which may alter the epithelial barrier property [Citation33]. Thus, it was evident from the results that intracellular uptake of hydrophobic drug (AM) could be enhanced by encapsulating in SLNs.
Elucidation of uptake mechanism of SLNs in Caco-2 cell monolayer
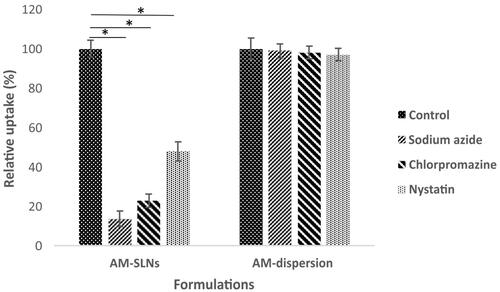
Various studies were conducted to elucidate the uptake mechanism of SLNs by Caco-2 cells. Sodium azide is reported to inhibit endocytosis [Citation17]. The results depicted that () only 13.68% ± 3.76% of AM from AM-SLNs was internalized in the presence of sodium azide by Caco-2 cells against control indicating endocytosis (carrier mediated transport) was involved in the uptake of AM-SLNs. Endocytosis process was further clarified in presence of other uptake inhibitors chlorpromazine (clathrin inhibitor) and nystatin (claveole inhibitor). The cellular uptake of AM-SLNs was reduced to 22.92% ± 3.41% in presence of chlorpromazine and 47.90% ± 4.98% in presence of nystatin respectively suggesting that clathrin as well as lipid raft/caveolae mediated endocytosis were involved in the uptake of AM-SLNs. However, the uptake of AM dispersion was not affected in presence of these inhibitors confirming the role of SLNs in facilitating absorption of AM through endocytosis.
Figure 8 Intracellular uptake of SLNs and drug dispersion in presence of specific inhibitors. *p < .05 compared to control group.
Transport study across Caco-2 cell monolayer
Transport study using transwell insert was performed to determine the permeation of formulations and drug dispersion across Caco-2 cells. It was observed that transport of AM was facilitated by AM-SLN as compared to drug dispersion as evident from Papp value of 6.25 × 10−6 cm/s and 0.82 × 10−6 cm/s, respectively. The AM-SLNs exhibited 7.62 times higher drug permeation than that of AM-dispersion at 4 h due to small size and large surface area and TPGS present in the SLNs [Citation34]. Moreover, the contribution of clathrin and claveole mediated endocytosis in uptake of SLNs may have increased the permeation of AM loaded SLNs as compared to drug dispersion. Thus, the enhancement of AM permeation by encapsulating in SLNs was verified in vitro using the Caco-2 cells as a model.
Oral BA study of AM-SLNs and AM dispersion
The results () exhibited marked difference between the pharmacokinetic profiles of AM-SLNs and AM dispersion. The peak plasma concentration (Cmax) after oral administration of AM-SLNs was found to be 1396.44 ± 116.81 ng/ml respectively which was 20.78 times that of AM dispersion (67.19 ± 8.40 ng/ml). The AUCtotal of AM-SLNs and AM dispersion was found to be 27460.50 ± 151.90 ng.h/ml and 547.12 ± 28.47 ng.h/ml respectively (). The AUC which denotes the extent of absorption was significantly higher for AM-SLNs than AM dispersion. The relative BA (Frelative) of AM-SLNs was 50.19-fold increased as compared to AM dispersion. Time to achieve maximum plasma concentration (tmax) for AM-SLNs and AM dispersion was 8.00 ± 0.21 h and 1.00 ± 0.31 h, respectively. This delay in tmax may be due to intracellular processing of lipids during lymphatic uptake [Citation35]. Compared with the AM dispersion, t1/2 and mean residence time (MRT) were prolonged with AM-SLNs owing to the slow elimination of AM from SLN. From these results, it can be concluded that intestinal absorption of AM was effectively enhanced by incorporating into SLN formulation compared with AM dispersion.
Figure 9 Plasma concentration versus time profile after oral administration of AM-SLNs and AM dispersion (n = 6). *p < .05 compared to drug dispersion.
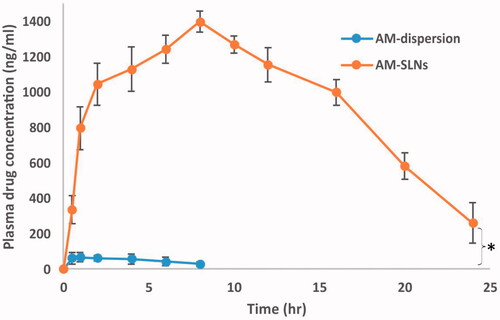
Table 1. Pharmacokinetic parameters of AM-SLNs and AM dispersion after oral administration.
Several mechanisms might be responsible behind BA improvement from AM-SLNs. (i) The particle size plays a pivotal role in increasing BA. Smaller sized particles can be efficiently uptaken by lymphoid tissues, where they bypass the first pass metabolism and increase BA [Citation19]. (ii) Increased effective surface of nanosized SLNs promote diffusion of drug and provide higher concentration gradient which increases rate of absorption. (iii) TPGS used in AM-SLN, is a lymphotropic excipient and has a role in enhancing chylomicron secretion [Citation36,Citation37]. It also has strongest inhibitory effect on CYP3A4 [Citation35] which also might have contributed in improvement of BA [Citation34]. (iv) It has been reported that pharmacokinetic profile of drug can be changed when incorporated into SLNs as the muco-adhesive property of SLNs can enhance the contact time with intestinal wall in vivo [Citation3,Citation38]. Thus, it can be concluded from the data that SLNs are a preferable carrier for the BA enhancement of AM, a lipophilic drug undergoing extensive first pass metabolism.
Lymphatic transport study of AM-SLNs using CHX
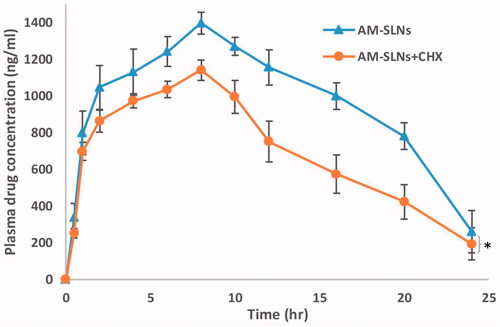
The data reflected that plasma concentration of AM in CHX treated rats was found to be lower than non-treated groups (). The peak plasma concentration of AM significantly reduced from 1396.44 ng/ml to 1140.34 ng/ml in rats after treatment with CHX. Moreover, the AUCtotal values reduced about 31.06% compared with that without treatment with CHX. Therefore, it can be said that the lymphatic pathway was associated in the uptake of AM-SLNs into the systemic circulation.
Figure 10 Plasma concentration versus time profile after oral administration of AM-SLNs to cycloheximide treated and non-treated (control) rats (n = 6). *p < .05 compared to control group.
Stability study
The stability of optimized AM-SLNs was monitored for 3 months at 2–8 °C and room temperature (RT) in terms of particle size, zeta potential and drug content (). No significant changes were observed in any of the assessed parameters when they were stored at refrigerated condition (p > .05).
Table 2. Characteristics of AM-SLNs after 3-months stability studies at different conditions.
However, particle size was increased whereas zeta potential and drug content were decreased when they were stored at RT. Drug content may have decreased at 30 °C because of drug expulsion from the lipid matrices at higher temperature [Citation39]. According to the DLVO theory, a system can be regarded as stable if the electrostatic repulsion dominates the attractive van der Waals forces. The particles have to overcome an energy barrier of electrostatic repulsion to approach closely and form agglomerates. If their velocity or kinetic energy is high enough they will collide. At high temperature (30 °C), the kinetic energy of a system increases which dominates the attractive forces over repulsive forces, reducing the zeta potential which may have led to particle aggregation [Citation40]. Hence, recommended storage condition for better stability of AM loaded SLN is under refrigeration.
Conclusions
In the present study, AM loaded SLNs with small particle size and high drug entrapment were successfully developed. The cell viability study against Caco-2 cells revealed that the formulation components and drug were non-toxic. The transport studies across Caco-2 cells showed that the uptake was energy as well as time dependent and endocytosis was mediated by both clathrinand claveolae routes. The permeability study showed that transport of AM across Caco-2 cell monolayer was significantly increased from AM-SLNs as compared to AM-dispersion. In vivo pharmacokinetic study showed significant enhancement in oral BA which was confirmed to be via lymphatic uptake pathway. Thus, it can be said that SLNs could be an effective strategy to augment oral BA of poorly soluble drugs.
Acknowledgements
The authors are grateful to Department of Science and Technology (DST), New Delhi, India, for providing ‘Innovation in Science Pursuit for Inspired Research (INSPIRE)’ Fellowship to Mitali Patel and to Alembic Pharmaceutical Limited, India for gift sample of asenapine maleate.
Disclosure statement
The authors declare no conflict of interest.
Related Research Data
References
- Stegemann S, Leveiller F, Franchi D, et al. When poor solubility becomes an issue: from early stage to proof of concept. Eur J Pharm Sci. 2007;3:249–261.
- Mishra B, Patel BB, Tiwari S. Colloidal nanocarriers: a review on formulation technology, types and applications toward targeted drug delivery. Nanomed Nanotech Biol Med. 2010;6:9–24.
- Harde H, Das M, Jain S. Solid lipid nanoparticles: an oral bioavailability enhancer vehicle. Expert Opin Drug Deliv. 2011;8:1407–1424.
- Zhang Z, Gao F, Bu H, et al. Solid lipid nanoparticles loading candesartan cilexetil enhance oral bioavailability: in vitro characteristics and absorption mechanism in rats. Nanomed Nanotech Biol Med. 2012;8:740–747.
- Minassian A, Young JW. Evaluation of the clinical efficacy of asenapine in schizophrenia. Expert Opin Pharmacother. 2010;11:2107–2115.
- Patel M, Sawant K. A quality by design concept on lipid based nanoformulation containing antipsychotic drug: screening design and optimization using response surface methodology. J Nanomed Nanotechnol. 2017;8:1–11.
- Das S, Ng WK, Kanaujia P, et al. Formulation design, preparation and physicochemical characterizations of solid lipid nanoparticles containing a hydrophobic drug: effects of process variables. Colloids Surface B. 2011;88:483–489.
- Soares S, Fonte P, Costa A, et al. Effect of freeze-drying, cryoprotectants and storage conditions on the stability of secondary structure of insulin-loaded solid lipid nanoparticles. Int J Pharm. 2013;456:370–381.
- Natarajan J, Baskaran M, Humtsoe LC, et al. Enhanced brain targeting efficacy of olanzapine through solid lipid nanoparticles. Artif Cells Nanomed Biotechnol. 2017;45:364–371.
- Sawant KK, Patel MH, Patel K. Cefdinir nanosuspension for improved oral bioavailability by media milling technique: formulation, characterization and in vitro-in vivo evaluations. Drug Dev Ind Pharm. 2016;42:758–768.
- Paliwal R, Rai S, Vaidya B, et al. Effect of lipid core material on characteristics of solid lipid nanoparticles designed for oral lymphatic delivery. Nanomedicine. 2009;5:184–191.
- Shi LL, Cao Y, Zhu XY, et al. Optimization of process variables of zanamivir-loaded solid lipid nanoparticles and the prediction of their cellular transport in Caco-2 cell model. Int J Pharm. 2015;478:60–69.
- Rajpoot K, Jain SK. Colorectal cancer-targeted delivery of oxaliplatin via folic acid-grafted solid lipid nanoparticles: preparation, optimization, and in vitro evaluation. Artif Cells Nanomed Biotechnol. 2018;46:1236–1247.
- Xiao B, Han MK, Viennois E, et al. Hyaluronic acid-functionalized polymeric nanoparticles for colon cancer-targeted combination chemotherapy. Nanoscale. 2015;7:17745–17755.
- Rivolta I, Panariti A, Lettiero B, et al. Cellular uptake of coumarin-6 as a model drug loaded in solid lipid nanoparticles. J Physiol Pharmacol. 2011;62:45–53.
- Jain A, Barve A, Zhao Z, et al. Comparison of avidin, neutravidin, and streptavidin as nanocarriers for efficient siRNA delivery. Mol Pharm. 2017;14:1517–1527.
- Shah MK, Madan P, Lin S. Elucidation of intestinal absorption mechanism of carvedilol-loaded solid lipid nanoparticles using Caco-2 cell line as an in vitro model. Pharm Dev Technol. 2015;20:1–9.
- Beloqui A, Solins M. n, Gascn AR, et al. Mechanism of transport of saquinavir-loaded nanostructured lipid carriers across the intestinal barrier. J Control Release. 2013;166:115–123.
- Parthasarathi TR, Srinivas TS, Sri MV, et al. Quantitative determination of asenapine maleate using reverse phase-high performance liquid chromatography. In J Pharm Bio Sci. 2012;3:360–366.
- Jose S, Anju SS, Cinu TA, et al. In vivo pharmacokinetics and biodistribution of resveratrol-loaded solid lipid nanoparticles for brain delivery. Int J Pharm. 2014;474:6–13.
- Memvanga PB, Coco R, Preat V. An oral malaria therapy: curcumin-loaded lipid-based drug delivery systems combined with β-arteether. J Control Release. 2013;172:904–913.
- Ravi PR, Aditya N, Kathuria H, et al. Lipid nanoparticles for oral delivery of raloxifene: optimization, stability, in vivo evaluation and uptake mechanism. Eur J Pharm Biopharm. 2014;87:114–124.
- Varshosaz J, Tabbakhian M, Mohammadi MY. Formulation and optimization of solid lipid nanoparticles of buspirone HCl for enhancement of its oral bioavailability. J Liposome Res. 2010; 20:286–296.
- Dahan A, Hoffman A. Evaluation of a chylomicron flow blocking approach to investigate the intestinal lymphatic transport of lipophilic drugs. Eur J Pharm Sci. 2005;24:381–388.
- Wang T, Shen L, Zhang Z, et al. A novel core-shell lipid nanoparticle for improving oral administration of water soluble chemotherapeutic agents: inhibited intestinal hydrolysis and enhanced lymphatic absorption. Drug Deliv. 2017;24:1565–1573.
- Yasir M, Sara UVS. Solid lipid nanoparticles for nose to brain delivery of haloperidol: in vitro drug release and pharmacokinetics evaluation. Acta Pharm Sin B. 2014;4:454–463.
- Almeida EDP, Costa AA, Serafini MR, et al. Preparation and characterization of chloroaluminum phthalocyanine-loaded solid lipid nanoparticles by thermal analysis and powder X-ray diffraction techniques. J Therm Anal Calorim. 2012;108:191–196.
- Carvalho SM, Noronha CM, Floriani CL, et al. Optimization of α-tocopherol solid lipid nanoparticles by central composite design. Ind Crops Prod. 2013;49:278–285.
- Venishetty VK, Chede R, Komuravelli R, et al. Design and evaluation of polymer coated carvedilol loaded solid lipid nanoparticles to improve the oral bioavailability: a novel strategy to avoid intraduodenal administration. Colloids Surfaces B. 2012;95:1–9.
- Nabi-Meibodi M, Vatanara A, Najafabadi AR, et al. The effective encapsulation of a hydrophobic lipid-insoluble drug in solid lipid nanoparticles using a modified double emulsion solvent evaporation method. Colloids Surf B Biointerfaces. 2013;112:408–414.
- Silva AC, Amaral MH, González-Mira E, et al. Solid lipid nanoparticles (SLN) – based hydrogels as potential carriers for oral transmucosal delivery of risperidone: preparation and characterization studies. Colloids Surf B. 2012;93:241–248.
- Liang M, Lin I, Whittaker MR, et al. Cellular uptake of densely packed polymer coatings on gold nanoparticles. ACS Nano. 2010;4:403–413.
- Sha X, Yan G, Wu Y, et al. Effect of self-microemulsifying drug delivery systems containing labrasol on tight junctions in Caco-2 cells. Eur J Pharm Sci. 2005;24:477–486.
- Chai GH, Xu Y, Chen SQ, et al. Transport mechanisms of solid lipid nanoparticles across Caco-2 cell monolayers and their related cytotoxicology. ACS Appl Mater Interfaces. 2016;8:5929–5940.
- Luo Y, Chen D, Ren L, et al. Solid lipid nanoparticles for enhancing vinpocetine's oral bioavailability. J Control Release. 2006;114:53–59.
- Luo CF, Yuan M, Chen MS, et al. Pharmacokinetics, tissue distribution and relative bioavailability of puerarin solid lipid nanoparticles following oral administration. Int J Pharm. 2011;410:138–144.
- Fan Z, Wu J, Fang X, et al. A new function of vitamin E-TPGS in the intestinal lymphatic transport of lipophilic drugs: enhancing the secretion of chylomicrons. Int J Pharm 2013;445:41147.
- Moghimipour E, Ameri A, Handali S. Absorption-enhancing effects of bile salts. Molecules. 2015;20:14451–14473.
- Ekambaram P, Hasan SAA. Formulation and evaluation of solid lipid nanoparticles of ramipril. J Young Pharm. 2011;3:216–220.
- Freitas C, Muller RH. Effect of light and temperature on zeta potential and physical stability in solid lipid nanoparticle (SLN™). Int J Pharm. 1998;168:221–229.