
Abstract
The present research seeks to investigate the process of mixing targeted gene delivery and transcriptional targeting. We have conjugated Polyethylenimine polymers (PEI) and molecules of poly (ethylene glycol). The next step was covalent attachment of anti-HER2 variables domains of camelid heavy chains antibodies (VHHs) or nanobodies (Nbs) to the distal terminals of NHS-PEG3500 in PEI-PEG nanoparticles. The whole procedure yielded PEI-PEG-Nb immunoconjugates. Having determined the properties of polyplexes, steps were taken to investigate the most efficient ratio of PEI polymers to pDNA molecules (N/P) so that the greatest rate of transfection may be obtained. This immune targeted nano biopolymer could condense the gene constructs that coded a transcriptionally targeted truncated –Bid (tBid) killer gene which was controlled by the breast cancer-specific MUC1 promoter. The favourable physicochemical properties matching both the size and zeta potential were observed in engineered polyplexes. Elevated transfection efficiency in HER2 positive cell lines using Nb-modified polyplexes were shown by the results of flow cytometry as compared against non-modified particles. 1.6 and 4.8 fold higher transfection efficiencies were observed in in vitro gene expression researches which used PEI-PEG-Nb/pGL4.50 compared to the situation when native PEI polymers were utilized in both BT-474 and SK-BR-3, respectively. A 2.22 and 3.62 fold rise in the level of tBid gene expression in BT-474 and SK-BR-3 cell lines relative to unmodified PEI treated cells was the result of transfection with PEI-PEG-Nb/pMUC1-tBid, respectively. In those HER2-positive cells which were transfected by targeted polyplexes, higher levels of cell death were observed. This fact points not only to the effective targeted delivery, but it is also indicative of transcriptional targeting efficiency of tBid killer gene when its expression is controlled by MUC1 promoter.
Introduction
The aim of cancer gene therapy is to induce cancer cell death with no severe damage caused to normal cells. Induction of apoptosis in cancer cells could be an endogenous mechanism for cell death. Apoptosis is mainly regulated by Bcl-2 family members. While there is general agreement that Bcl-2 family members, including Bcl-2 itself, Mcl-1, Bcl-x and Bcl-w, can inhibit apoptosis, provide clonogenic protection and act upstream of the mitochondria, how they work exactly is not known [Citation1]. Bid pro-apoptotic protein is a BH3-domain-only subgroup of BCL-2 family member which is capable of binding to either BCL-2 or BAX and inducing cell death. It has been demonstrated that cytosolic 22 kDa BID protein is activated by caspase-8 cleavage which in turn is activated by death signals [Citation2–4]. The resulting 15 kDa molecule, which is recognized as a truncated p15 BID (tBID), is translocated to the mitochondrial outer membrane, where tBID promotes the release of cytochrome c from mitochondria [Citation3,Citation4] and activates the intrinsic pathway of apoptosis and ultimately leads to cell death. tBid is a suitable pro-apoptotic/killer gene due to its proper size and because it doesn’t require any precious modification to become fully active [Citation5].
Viral vectors are the most efficient gene delivery agents; but their application in gene therapy is restricted due to their relatively high immunogenicity, risk of mutagenesis, and other safety concerns [Citation6]. Non-viral gene delivery agents exhibit several advantages over viral vectors; including their ability of specific targeting, incapability of integrating into the host genome, low immunogenicity, unlimited size of DNA to be transfected, and ease of production/synthesis. Among non-viral vectors, positively charged polyethyleneimine(PEI), a synthetic polymer, is most extensively evaluated due to its high DNA condensing and transfection efficiency [Citation7,Citation8]. PEI polymer, due to its large number of primary functional surface amine groups, is suitable for coupling of targeting ligands, including small peptides or antibodies. Positively charged nano-carriers such as PEI usually demonstrate higher cell surface binding and internalization abilities because of their negatively charged cell surface [Citation9,Citation10]. On the other hand, this positive charge leads to unspecific cellular binding and fast blood clearance. In order to overcome PEI toxicity, its stability in blood circulation, and protection of the condensed DNA, the positive surface charge of PEI/DNA polyplexes should to be masked, which can usually be achieved by grafting the hydrophilic polymers such as poly (ethyleneglycol) (PEG) to PEI, the so-called process of PEGylation [Citation11–14]. Although PEGylation enhances the biocompatibility of PEI polycation; however, it decreases transfection ability of this nanoparticle. In order to overcome this obstacle, targeting ligands could be included into PEI-PEG copolymer to enhance their specificity and gene transfection ability [Citation13–15].
One of the receptors used for tumour targeting is HER2 because it has a limited expression on normal cells; however, high levels of its expression are observed in 25–30% of human breast carcinomas and other types of cancer observed in humans. Major actors in the process of HER2 targeted gene delivery are monoclonal antibody grafted polyplexes which are directed against HER2. There is no doubt that high levels of antibody’s affinity to a particular target results in a selective form of binding. The smallest intact domain from heavy-chain-only antibodies which naturally occur in Camelidae, those bind antigens are VHHs or nanobodies (Nbs) [Citation16,Citation17]. Properties such as high affinity, specificity, homology to the human family of VH3 and low immunogenicity of nanobodies along with their nanometric size are factors that render them favourable for targeting antigens in obstructed locations, such as tumours.
An anti-HER2 Nb (RR4) was recently chosen from a phage display library whose flow cytometry analysis pointed to the fact that this Nb can specifically localize on the surface of plasma membrane of those positive cell lines that belong to HER2. These observations were a proof of its targeting ability [Citation18].
Using transcriptional targeting strategy along with HER2 targeting by anti-HER2-specific Nbs was an effort to limit tBID expression to those breast cells diagnosed with HER2-positive cancer. MUC1 gene promoter was utilized for transcriptional targeting as its tumour specificity and elevated protein expression capabilities in breast cancer had been confirmed [Citation19]. The present research utilized anti-HER2 targeted PEI nanoparticle as compacting agents for transcriptionally targeted tBid killer gene construct. The next step was evaluation of their selective delivery into HER2 expressing cell lines. This research seeks to prove the concept of combining Nb-based targeting with transcriptional targeting as a safe and efficient alternative method to deliver transgenes to certain cells in vitro.
Materials and methodology
Materials
Sigma-Aldrich Company (St Louis, MO) provided our research with branched 25 kDa PEI, cysteine hydrochloride and 2, 4, 6-trinitrobenzene sulfonic acid (TNBSA or TNBS) reagent, while Jenkem Technology (Beijing, China) provided the NHS-PEG3500-MAL required for our project. 2-Iminothiolane-hydrochloride (Traut’s reagent) and 5, 5′-Dithio-bis-2-nitrobenzoic acid (Ellman’s reagent) were two other materials required for this research obtained from Pierce (Rockford, IL). Other analytical grade reagents used in this study were bought from Sigma-Aldrich Company. The Cell lines were bought from DSMZ (Braunschweig, Germany).
Cell lines
Two HER2 overexpressing human breast cancer cell lines, BT-474 and Sk-Br-3 cells were used as HER2 positive cell lines, whereas MCF-10A (human non-tumourigenic mammary epithelial cell line) was used as normal non-cancerous breast cancer cell line. All cell lines were obtained from DSMZ (Braunschweig, Germany). The SK-BR-3 cell line was cultured in DMEM (Gibco/Invitrogen, Carlsbad, CA) media supplemented with 15% fetal bovine serum (FBS), 100 U/100 μg ml−1 penicillin/streptomycin and 365 μg/ml l-glutamine. MCF-10A cell line was cultured in DMEM/F-12 (1:1) (Gibco/Invitrogen) media supplemented with 5% donor horse serum, 365 μg/ml l-glutamine, 20 ng/mL epidermal growth factor (EGF), 10 µg/mL insulin, 0.5 µg/mL hydrocortisone and 100 U/100 μg ml−1 penicillin/streptomycin at 37 °C in a 5% CO2-humidified incubator. BT-474 cells were maintained in RPMI-1640 (Gibco/Invitrogen) supplemented with 20% (for BT-474 cell line) FBS and 100 U/100 μg ml−1 penicillin/streptomycin.
Gene constructs
A modified version of the pNIC28-DsbA1 vector was used in order to clone DsnA2-RR-4 where the researchers decided to neglect the export signal sequence. The above-said vector is responsible for encoding DsbA (periplasmic protein disulfide isomerase I). Next, you could observe a sequence of an N-terminal hexahistidine tag and a TEV protease recognition site (amino acid sequence ENLYFQ*S) and two unique BsaI sites for ligation-independent cloning (LIC). Utilizing standard ligation-independent cloning (LIC) protocols [Citation20], those DNA sequences which encoded the proteins of interest flanked by LIC sites were PCR amplified, T4 polymerase-treated and cloned into expression vectors. A mismatch was designed in TEV P2’ position so that it could be mutated to a serine. LB agar plates that contained 50 μg/ml kanamycin and 5% sucrose were utilized to select those vectors containing the insert. The sequences of plasmids were verified before they were transformed into expression strain BL21 (DE3)-R3-pRARE2.
tBid gene was amplified by PCR on peripheral lymphocytes cDNA and cloned in the pCDNA3.1/Hygro+ (Invitrogen, Carlsbad, CA) downstream of the CMV promoter to pave the way for pCMV-tBid construction. The other promoter amplified by PCR on lymphocyte genomic DNA used to replace the CMV promoter was MUC1 promoter (pMUC1) which resulted in the production of pMUC1-tBid.
In the process of tBid control vector designing, CMV promoter was replaced by synapsin I promoter (pSyn) so that we may obtain pSyn-tBid. As for a strong eukaryotic promoter, a breast cancer-specific promoter, and a promoter specific for central nervous system, pCMV, pMUC1 and pSyn were selected respectively [Citation21].
Synthesis and characterization of PEI-PEG and PEI-PEG-Nb conjugates
25 kDa bPEI polymer in 2 ml of degassed phosphate buffer (pH 7.5) was mixed with bi-functional PEG (NHS-PEG3500-Mal) with a molar ratio of 10:1 (PEG: PEI polymer) at room temperature (RT) under nitrogen for 2 h in order to reactivate bi-functional PEG. Amicon ultrafilters (MWCO 10 kDa, Millipore, Schwalbach, Germany) was used in order to separate free unbound PEG molecules by ultrafiltration. Based upon TNBS assay and maleimide assay with Ellman’s reagent, we managed to determine the extent of PEG conjugation [Citation22].
Nanobody molecules were thiolated using Traut’s reagent, with nanobody to Traut molar ratio of 10:1 was the first step taken to conjugate molecules of nanobody to maleimide distal end of PEGylated PEI copolymers [Citation23]. Utilizing Amicon ultrafiltration, the unbound Traut molecules were removed by ultrafiltration (MWCO 30 kDa). The next step was adding Nb to PEI-PEG at a molar ratio of 2:1 so that it may be allowed to react for 18 h under nitrogen. Using Amicon ultrafilters (MWCO 50 kDa), the final conjugates were removed from any unbound Nb and PEI molecules. By coupling Trastuzumab monoclonal antibody to PEGylated PEIs, we managed to obtain PEI-PEG-Trastuzumab conjugates. The whole process was similar to what was observed in the synthesis of PEI-PEG-nanobody conjugates but the molar ratio set for Traut reagent to Trastuzumabzumb antibody was (20:1) and when the process of thiolation was over, Amicon ultrafilter (MWCO 50 kDa) removed the unconjugated Traut molecules. Amicon ultrafilter (MWCO 100 kDa) separated the resulting conjugates from unbound antibodies. To determine the properties of conjugates, proton nuclear magnetic resonance (1H-NMR) utilizing deuterated water (D2O) and Bruker 400 MHz Avance II + NMR spectrometer (Bruker, Rheinstetten, Germany) were utilized.
Polyplex formation
Addition of an equal volume of pDNA solution to an equal amount of various PEI formulations in order to get a favourable ratio of N/P helped us prepare polyplexes. Before incubating polyplexes for 30 min at room temperature prior to their use, they underwent a process of vortexing for half a minute.
Gel retardation assay
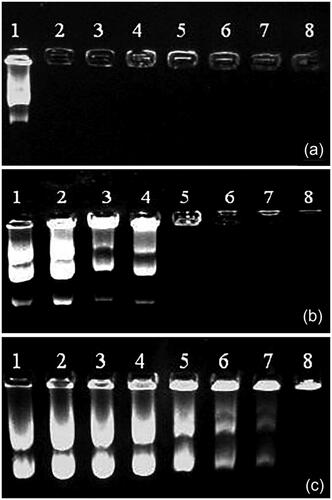
In order to confirm DNA complexation and condensation, we utilized agarose gel retardation assay in the presence of ethidium bromide in 1 M Tris-acetate-EDTA (TAE) buffer solution. PEI, PEI-PEG and PEI-PEG-nanobody copolymers which were mixed with 1 μg pDNA underwent gel retardation assay at an N/P ratio of 6. As much as 1 μg free pDNA was added to the gel as a controlling factor.
Particle size and zeta potential measurement
The size and zeta potential of polyplexes were measured using Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). Based upon the method used for gel retardation assay experiments, steps were taken to prepare polyplexes. For dilution of solutions, 800 μl of MilliQ-water was used.
In vitro toxicity evaluation of nanocarriers
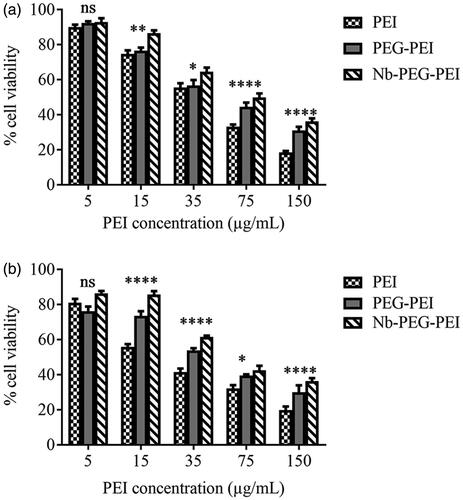
Cytotoxicity of PEI, PEG-PEI and Nb-PEG-PEI carriers was evaluated by 3–(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay on two cell lines (BT-474 and MCF-10A). The cells were seeded in 96-well plates at an initial density of 10,000 cells/well, 24 h prior to in vitro cell viability assay and then incubated at 37° C with different concentrations of PEI polymer or derivative copolymers (0, 5, 10, 15, 35, 75 and 150 μg/ml) without gene loading in presence of fresh media. All experiments were performed in triplicate. After cell incubation for 4 h in the presence of polymer or copolymers, the medium was removed and replaced with fresh medium containing FBS and incubated at 37° C for 48 h. Then MTT reagent was added to the cells and the plates were incubated in the incubator for 4 h at 37° C. Then the supernatants were removed gently, and the formazan crystals were solubilized in 100 µl DMSO and the colour intensity was assessed by spectrophotometry at 570 nm. Cells without treatment (100% viability) and cells without the addition of MTT (as blank) were also included as control.
Optimizing DNA concentration and N/P ratio
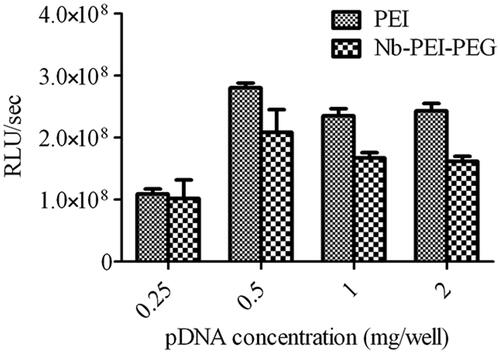
Luciferase (Luc) reporter assay was used to determine the optimal amount of pDNA for transfection. To achieve this, 25 kDa bPEI and PEI-PEG-nanobody polymers were mixed with 0.5, 1 and 2 μg levels of pGL4.50 (Luc-encoding plasmid). Before transfecting polyplexes, they underwent the process of vortexing for half a minute and incubation for half an hour in room temperature. All polyplexes were fresh, i.e. they were prepared exactly before use. When the most efficient level of pDNA was determined, subsequent experiments were designed and planned based upon the above-mentioned amount. Transfections were conducted utilizing enhanced green fluorescent protein (EGFP) and Luc in order to encode pDNA, then the optimal charge ratio of PEIs/DNA (N/P ratio) for gene delivery experiments was calculated. One day before transfection, as much as 1 × 104 of BT-474, SK-BR-3, and MCF-10A cells were planted in plates with 96 wells. On the day chosen for transfection, as much as 0.5 μg pGL4.50 was mixed with PEI polymers at four different N/P ratios of 2, 4, 6 and 10. The resulting mixture was then incubated for 30 min at room temperature and added to cells in serum-free media. Four hours later, a serum containing the medium was used to change the medium. The media was separated after 30 min, then PBS solution was used to wash the cells which were treated with 40 μl of luciferase cell culture lysis reagent (Promega). Half an hour later, a mixture of 20 μl of cell lysates and 100 μl luciferase assay reagent (Promega) was prepared. It was then immediately analyzed using LUMO luminometer (Autobio Labtec Instruments Co., Zhengzhou, China). According to the specific program designed for our research, the microplate reader performed a 2-s measurement delay and then a 10-s measurement read for luciferase activity as it was demonstrated in the manual. OD280 measurements by Shimadzu UV-240 UV-Vis spectrophotometer were utilized to quantify the protein content in cell lysates. Luciferase activity was reported as RLU/mg protein. For negative control, untransfected cells and those cells transfected with naked pDNA were used. A procedure similar to the one observed in Luc gene expression was followed for EGFP expression studies. Observation of cells was accomplished by an inverted fluorescence microscope and the efficacy of transfection was qualitatively assessed 48 h after transfection. The most efficient N/P ratio for each cell line was used for subsequent experiments.
In vitro Luc gene expression analysis
Twenty-four hours before transfection, BT-474, SK-BR-3, and MCF-10A Cell lines were seeded at a density of 1 × 104 per well of 96-well plates. A mixture of PEI, PEI-PEG, PEI-PEG-Nb and PEI-PEG-Trastuzumab with 0.5 μg of pGL4.50 was then prepared and used for transfection studies. The very same instructions introduced above were followed. Luciferase activity was measured 30 h after transfection. Every treatment was repeated three times.
In vitro tBid killer gene expression
Twenty-four hours before transfection, BT-474, SK-BR-3 and MCF-10A cells were all seeded at a density of 1 × 104 cells/well of 96-well plates in complete medium. The cells were stored in antibiotic and serum free media for 2 h before transfection. A mixture was prepared using PEI, PEI-PEG, PEI-PEG-NB and PEI-PEG-Trastuzumab with 0.5 μg of pMUC1-tBid. The resulting mixture was then added to the wells. In a similar experiment conducted to evaluate transcriptional targeting of the tBid gene, three different promoters including pCMV, pMUC1 and pSyn I were utilized. Transfection experiments were carried out using Nb-PEG-PEI polymers mixed with as much as 1 μg of either pEGFPN1, pCMV-tBid, pMUC1-tBid or pSyn-tBid. Expression of tBid gene in terms of its killing activity was measured 2 days after transfection using LDH cytotoxicity assay for both experiments.
Co-transfections and reporter assays
In order to determine the killing efficiency of tBid killer gene under the control of different promoters, it is necessary to measure the luciferase reporter activity of the pGL4.50 plasmid utilized in co-transfections experiments mixed with tBid-expressing constructs. Exactly 24 h before carrying out transfection, cells were seeded at a density of 1 × 104 cells per well of 96-well plates. Preparation of polyplexes required 250 ng tBid-expressing plasmids or pCDNA3.1 vector (without tBid gene) along with 250 ng of pGL4.50 (Luc-expressing plasmid) condensed with PEI/PEI-PEG-Nb. Luciferase activity in tBid- transfected cells relative to pcDNA3.1-transfected cells was estimated 30 h after transfection luciferase reporter activity.
Statistics
GraphPad Prism 6.0 was utilized to conduct statistical analyses. The results were presented in the form of mean ± standard deviation (SD) of three samples. In order to assess the statistical differences between the group means, the authors decided to utilize students’ unpaired t-test and 2-way ANOVA with Tukey correction.
Results and discussion
Synthesis and determining the properties of PEI-PEG and PEI-PEG-Nb conjugates
The fact that PEGylation of polymers with the molar ratio of 10/1 (PEG/PEI) yielded conjugates containing an average of 21 PEG chains per PEI molecule was confirmed by TNBS and Ellman essays. An average of 1.5 thiol groups per nanobody molecule on the basis of Ellman assay was introduced by Thiolation of nanobody. UV measurement at 280 nm arrived at the following results: final nanobody-functionalized polymers had ∼0.48 nanobody per PEI-PEG molecule and the conjugation rate was about 49%. The overall polymer yield after purification was about 63%, as determined by the TNBS assay. PEI-PEG-Nb conjugates were further characterized by 1H-NMR spectroscopy (400 MHz) as exemplified in . 1H-NMR spectra revealed multiple peaks corresponding to the –CH2– group of PEI at δ2.78–3.17. The successful conjugation of PEG molecules was shown by the proton signals between 3.41 and 3.62 ppm, which belonged to (–CH2CH2O–) units of PEG chains. Nanobody conjugation was confirmed by proton signals transmitted between 6.7 and 8 ppm. They showed evidence of benzene rings of atomic amino acids observed in sequences of nanobody protein.
Figure 1. 1H-NMR spectrum of Nb-PEG-PEI conjugates (D2O as the solvent). The insets are expanded images of 0.5–4 and from 5 to 9.5 of the spectra.
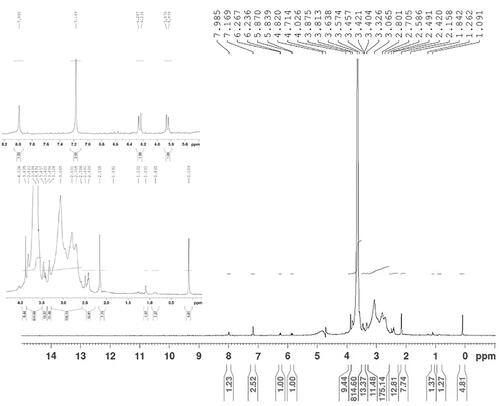
Figure 2. Agarose gel electrophoresis of (a) PEI, (b) PEG–PEI, and (c) Nb–PEG–PEI polyplexes. Lanes 1–8 represent N/P ratios of 0 (control, pDNA only), 0.25, 0.5, 1, 2.5, 5, 7.5, and 10, respectively.
Agarose gel retardation assay
The fact that PEI could efficiently retard DNA at N/P ratio of 0.5 and higher was confirmed by Agarose gel electrophoresis retardation assay (). We also observed effective complex formation with PEI-PEG at N/P ratio of 2.5 and higher and PEI-PEG-Nb polymers mixed with pDNA at N/P ratio of 10. Upon observation of results, the researchers came to realize that polymers complexation with pDNA was influenced by conjugation of PEI-PEG with nanobody molecules and adding these targeting moieties could influence the tendency of polymers to bind with pDNAs [Citation21,Citation24].
Particle size and zeta potential assessment
In order to gain a better understanding of the size and electrophoretic properties of the resulting conjugates, Dynamic Light Scattering (DLS) and zeta potential measurements were conducted. points to the fact that the hydrodynamic diameter decreased from 158.2 ± 7.2 for PEI-PEG molecules to 123.4 ± 7.4 for Nb-PEG-PEI molecules.
Table 1. Hydrodynamic diameter and zeta potential of the polyplexes formed by pEGFPN1 and unmodified, PEGylated and nanobody conjugated PEI polymers (N/P = 10).
This phenomenon can be explained by the fact that peptide or protein conjugation can decrease the aggregation of PEGylated PEIs [Citation25]. Measurement of zeta potential pointed to +13.7 ± 1.3 and +4.2 ± 1.8 as the charge values of PEI-PEG and PEI-PEG-Nb, respectively. These results point to a rather neutral charge situation for the final PEI-PEG-Nb structure. The other conclusion derived from these results is that the positive charges of the free amines on the PEI surface cannot be thoroughly detected and this fact is probably due to the shielding capacity of the bigger structure of nanobody moieties on the surface of the final conjugate. The presence of PEG and nanobody moieties prevents formation of bigger aggregates in the solution, although the very low surface charge encourages expectation of larger particles from the PEI-PEG-Nbconjugates.
Cytotoxicity assessment of gene nanocarriers
In order to investigate the influence of PEG grafting on in vitro cytotoxicity of PEI polymer and then the influence of Nb conjugation on cytotoxicity of PEG-PEI copolymer, viability of BT-474 and MCF-10A cell lines was evaluated by using MTT assay after incubating the cells with different concentrations of PEI, PEG-PEI and Nb-PEG-PEI such as 5 µg/mL, 15 µg/mL, 35 µg/mL, 75 µg/mL and 150 µg/mL for 4 h. Cell viability was calculated relative to the MTT value for untreated cells taken as 100% viability of cells. Based on our findings, by increasing the polymer concentration, the cell viability was decreased. As demonstrated in , at lower concentrations, all PEI derivatives were almost non-toxic. It was observed that at the highest carrier concentration (150 µg/mL), Nb-PEG-PEI could decrease the toxicity of PEI in both cell lines by two folds. It was also demonstrated that PEGylated PEI showed lower toxicity toward cell lines than non-PEGylated polymer which can be attributed to the shielding effect of PEG chains on the positive charges of PEI polymer.
Figure 3. Cytotoxicity evaluation of PEI, PEG-PEI and Nb conjugated PEG-PEI nanocarriers at various concentrations of 5, 15, 35, 75 and 150 µg/ml in BT-474 (a) and MCF10A (b) cell lines. Untreated cells were used as negative controls. Data are expressed as mean ± standard deviation (n = 3); ****, ***, **, *, and ns (not significant) indicate p < .0001, .001, .01, .05, and p > .05 when Nb conjugated PEG-PEI compared with unmodified PEI polymer at the same concentration.
How DNA concentration and N/P ratios influence gene delivery
The effect of DNA concentration on transfection efficiency of polyplexes was studied thoroughly using 0.25, 0.5, 1 and 2 μg pGL4.50 (plasmid encoding Luciferase reporter gene) combined with unmodified and Nb-modified polymers. The previously described processes were followed to prepare polyplexes. According to what is observed in , using 0.5 μg pDNA for transfection yielded the best transfection efficiency. Following this observation, later experiments utilized 0.5 μg of plasmid DNA.
Figure 4. Influence of plasmid DNA amount on luciferase reporter gene expression. An amount of 0.25, 0.5, 1 and 2 μg pGL4.50 were mixed with PEI and Nb-PEG-PEI with constant N/P ratio of 10.
PEI polymers were mixed with pEGFPN1 and pGL4.50 at four N/P ratios of 2, 4, 6 and 10 and used to transfect BT-474, SK-BR-3 and MCF-10A cells so that the optimal charge ratio of polymer/DNA on polymer-mediated gene delivery can be determined. As the GFP expression and Luciferase activity indicated, no significant difference was observed in transfection efficiency of polyplexes in various cell lines at a defined N/P ratios.
The peak of transfection efficiency was observed at N/P ratio of 6 for all cell lines ( and ). The transfection activity in all different cell lines was increased as the N/P ratio increased from 4 to 6. Lower transfection ability in all cell lines was the outcome of increasing N/P ratio over the specified levels. Experiments with both unmodified and modified polymers were conducted based upon this N/P ratio. Similar observations for PEI polyplexes were also reported by Boussif et al [Citation7] and Oh et al [Citation26].
Figure 5. Influence of the N/P ratio of PEI polymers complexed with pGL4.50 plasmid on luciferase reporter gene expression. (N/P ratio of 2, 4, 6 and 10 were used) in SK-BR-3 (a), BT-474 (b) and MCF10A (c) cell lines.
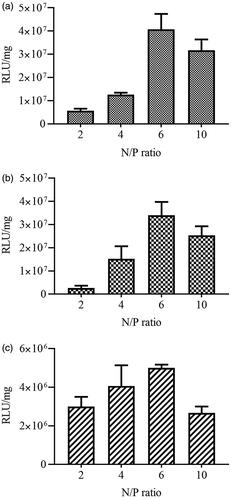
Figure 6. Results of N/P ratio optimization. Polyplexes were prepared by mixing pEGFPN1 plasmid and PEI polymers at various N/P ratios of 2, 4, 6 and 10. Transfection efficiency was determined 48h post transfection using fluorescence microscopy imaging. The micrographs (a), (b) and (c) show the cells which transfected with N/P ratio of 2; (d), (e) and (f) show the cells which were transfected with N/P ratio of 4; micrographs (g), (h), and (i) show the cells which were transfected with N/P ratio of 6 and (j), (k) and (l) show the cells which were transfected with N/P ratio of 10.
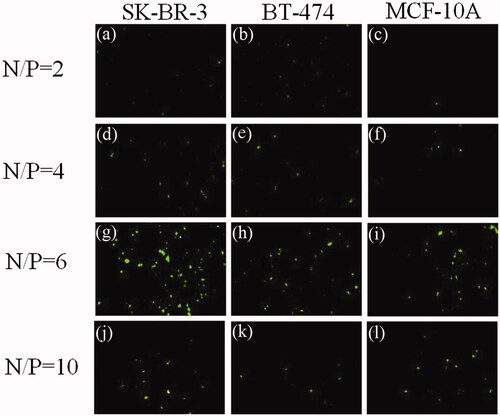
In justification of this phenomenon, one may point to this fact that at lower N/P ratios, PEI polyplexes incline to aggregate and compose larger nanoparticles. A rise in the level of N/P ratio may result in smaller nanoparticles which facilitate cellular uptake via endocytosis.
In vitro gene expression analysis
Gene transfection was studied in vitro through detection of Luciferase (reporter gene) and expression of tBid (killer gene) in HER2-positive and HER2-negative cells and they were compared against PEI/pDNA and PEI-PEG/pDNA polyplexes so that the potential of using PEI-PEG-Nb/pDNA polyplexes in targeted tumour therapy could be evaluated. To achieve this end, various formulations of polymers (unmodified, PEGylated and targeted) were utilized to deliver pGL4.50 and pMUC1-tBid gene constructs to cells.
represents the luciferase expression results of various cell lines which are transfected by polyplexes that contain pGL4.50. The first phase of researches conducted on gene delivery pointed to the fact that the highest expression of luciferase among all polyplexes was observed in Trastuzumab-modified polyplexes which were utilized for transfection of HER2-positive cell lines. The transfection efficiency obtained from Nb-tagged polyplexes was 1.58 and 1.36 times higher than what was actually observed in unmodified PEI in BT-474 and SK-BR-3 cells, respectively. Neither Trastuzumab nor nanobody-conjugated polyplexes could not cause a significant rise in transfection of HER2-negative cell line (MCF-10A). This observation can probably be justified on the ground of more pDNA entry into HER2-positive cells by targeted nanoparticles.
Figure 7. Luciferase gene construct pGL4.50 in SK-BR-3 (a), BT-474 (b) and MCF10A (c) cell lines transfected using PEI, PEI-PEG, PEI-PEG-Nb and PEI-PEG-Trastuzumab vectors.
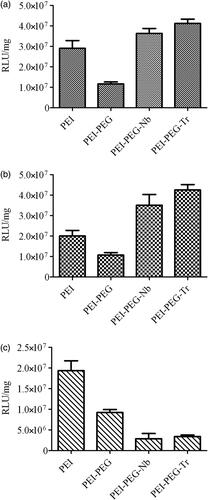
The second phase of gene delivery researches utilized pMUC1-tBid gene construct. The results of the gene delivery achieved through those polyplexes that contained this construct were indicative of the fact as a result of treating cell lines by PEI-PEG polyplexes, a decrease in expression of tBid gene in comparison with cells treated with unmodified PEI polyplexes was led to less cell death in PEI-PEG treated cells as the result of cell viability measurement, than those treated with PEI (). This phenomenon can probably be justified by the lower entry of PEGylated PEI polyplexes. Compared to PEI polymers, tBid gene expression using anti-HER2 nanobody-conjugated polyplexes was higher in BT-474 and SK-BR-3 cell lines. This issue causes a further rise in cell death compared to what was observed for HER2-negative cells. The highest expression of tBid gene and therefore the least amount of cell viability in HER2-positive cells was observed in Trastuzumab modified polymers. These observations were completely in line with luciferase expression experiments. As a result, targeting HER2-positive cell lines with Trastuzumab-modified polyplexes yielded a dramatic rise in cell death upon the killer gene efficient entry and expression in the cells. As expected, lower levels of specificity toward low/ non-HER2 expression cell line (MCF-10A) was observed in targeted polyplexes. As a justification for this observation, one may point to the fact that tBid expression level and cell viability were comparable with PEG-PEI treated MCF-10A cell line.
Transcriptional targeting activity of breast cancer-specific promoters
The promoter of MUC1 gene (pMUC1), whose effective specificity for breast cancer cells was previously discussed [Citation25,Citation26], was selected to undertake the transcriptional targeting of tBid gene expression to cancer cells. As it has been proven that CMV promoter is one of the most powerful promoters in almost all eukaryotic cells (utilized as a positive control) and as Synapsin I promoter yields targeted transcription of desired genes in central nervous system (utilized as a negative control), these two promoters along with MUC1 promoter were utilized to evaluate the transcriptional targeting efficacy. As it is indicated in and , cell death analysis was used to evaluate the transcriptional activity of these promoters in different cell lines. In line with our predictions, CMV promoter was shown to be the most efficient and active promoter in all tested cell lines, although no significant difference between the levels of promoter activity was observed in BT-474 and SK-BR-3 cells treated by CMV and MUC1 promoter constructs. Some extent of cell death was caused by the leak of tBid expression in HER2 negative cells. However, more restricted expression of tBid killer gene to HER2 positive breast cancer cell lines was mainly the incorporation of anti-HER2 nanobodies on polymers’ surface that caused the enhanced cellular uptake of polyplexes in these HER2-positive cell lines.
Figure 8. Viability of cells treated with a different pMUC1-tBid gene containing polyplexes. Viability percent of transfected and non-transfected SK-BR-3 (a), BT-474 and MCF10A (c) cells were evaluated using MTT assay.
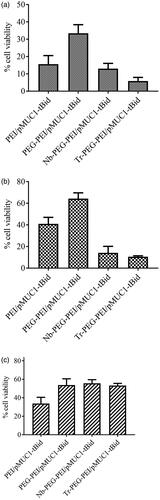
Figure 9. Viability of SK-BR-3 (a), BT-474 (b) and MCF10A (c) cells treated with PEI or Nb-PEG-PEI complexed with pEGFPN1, pCMV-tBid, pMUC1-tBid and pSyn-tBid. Viability percent was evaluated using LDH assay which was done as triplicate. Untreated cells and 100% lysed cells were used as negative and positive controls, respectively.

As of MUC1 promoter activity, the following order was observed in the highest values of tBid expression: SK-BR-3 > BT-474 > MCF-10A cell lines. The interesting point was that considering targeted-polyplexes containing pSyn-tBid, cell viability measurements in all cell lines remained comparable with unmodified polyplexes. The high activity of MUC1 promoter in both BT-474 and SK-BR-3 served to prove that combining HER2 targeted delivery with transcriptional targeting was a safe way to restrict transgenes expression to HER2-positive breast cancer cells.
No significant cell death was caused by PEI-PEG-N/pEGFPN1 polyplexes used in this experiment. These results were a strong proof that nanobody molecules’ capability of recognizing HER2 antigen could not affect cell viability. It also introduced the killing efficiency of tBid protein as the main cause of cell death observed in all experiments. These results are in line with the previous data showing the killing efficiency of tBid killer gene under the control of the MUC1 promoter following transfection of MUC1 over-expressing cancer cells by using Lipofectamine 2000™ [Citation27].
Co-transfection experiments
The co-transfection experiments seek more thoroughly to study the activity of tBid gene under the control of CMV, MUC1 and Syn promoters. Here, tBid gene bearing constructs were used for cell transfection under the control of different promoters together with a luciferase reporter gene expressing plasmid. These experiments utilized PEI-PEG-Nb and unmodified PEI polyplexes. The low transfection efficiency of cell lines, which could result in an inaccurate interpretation of cell death measured by LDH assay was the main motif for selecting this approach.
Assuming a 100% viability rate for luciferase transfected cultures which were included in the test as control, the researchers calculated the percentage of cell survival by dividing the luciferase activity of tBid-transfected cultures to luciferase-transfected cultures. According to cell viability results presented in , the most efficient combination for killing cells was tBid killer gene expression under the control of CMV promoter. Using Nb-modified polyplexes contributed to higher killing efficiency. According to , a decrease from 50% and 55% (using PEI) to 32 and 38% (using Nb-PEI-PEG) was observed in the cell viability of the cells-co-transferred by pCMV-tBid/pGL4.50, for BT-474 and SK-BR-3 cells respectively. However, a decrease of 41% (using PEI) to 18% (using Nb-PEG-PEI) was observed in normal breast cell lines with a low level of HER2 expression. This observation for MCF10A cell line could be explained by low levels of HER2 expression on the surface of these cells which lead to the lower binding of Nb-PEG-PEI nanocarriers to these cells compared to PEI carrier and thus lower entry of its gene loading.
Figure 10. Viability assessment of SK-BR-3 (a, d), BT-474 (b, e) and MCF10A (c, f) cells co-transfected with tBid containing constructs with Luciferase bearing plasmid. PEI (a, b, c) and Nb-PEG-PEI (d, e and f) polyplexes were used for co-transfection experiments.
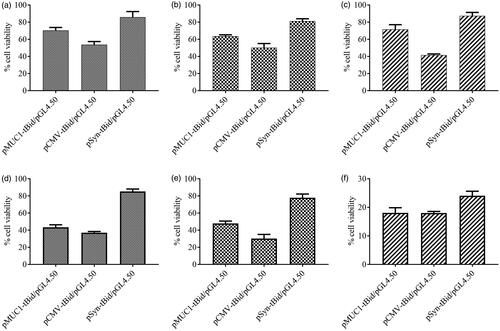
The fact that MUC1 promoter could also mediate cell death in almost all cell lines used even MCF10A, may be explained by the low level of MUC1 gene expression in this cell line. A 1.5 fold rise of cell death in both BT-474 and SK-BR-3 cell lines compared to unmodified polyplexes was observed when tBid expression was targeted to breast cancer cell lines with Nb-modified polyplexes carrying pMUC1-tBid gene construct. However, transfection of pMUC1-tBid to them by nanobody-modified polyplexes resulted in lower levels of the survival rate of MCF-10A cells. This can be justified on the ground of less entry of these polyplexes and could be explained by the lower presentation of HER2 on these cells’ surface and so less entry of their loading plasmids. Therefore however, less cytotoxicity due to MUC1-tBid could be expected in this cell line, but on the other hand because of less entry of luciferase gene bearing plasmid, lower luciferase signal was produced by these cells and due to this fact that the cell viability was calculated from luciferase activity in this test, the estimated cell viability was lower.
No significant tBid expression level was shown by Synapsin I promoter in breast cancer cell lines and all the pSyn-tBid-transfected HER2 overexpressing cells with both unmodified and nanobody-modified polyplexes were almost viable compared to untreated control cells. In normal breast cell line, it was observed that cell viability significantly decreased by using nanobody-modified polyplexes, this finding could be justified by lower expression of HER2 antigen on the surface of these cells and therefore fewer introductions of the polyplexes into them.
According to Kazhdan et al., the MUC1 promoter generally showed higher levels of efficiency in directing the cell killing, especially in breast cancer despite significantly low expression of genes as a result of transient transfection. They also reported the expression of a high level of MUC1 protein by MCF-10A normal mammary epithelial cells. The result was relatively high levels of MUC1 promoter activity in these cells [Citation19].
tBid expression driven by MUC1 promoter could mediate cell death relatively lower than CMV promoter but, the killing efficiency was transcriptionally targeted and specified to breast cancer cells as it was unable to achieve a significant cell death in MCF10A (a normal breast cell line considered as a non-cancerous cell line with low levels of MUC1 and HER2 expression). However, relatively same levels of cell death could be caused by it in all cell lines measured by luciferase activity when using PEI polymer as a gene carrier. On the contrary, higher levels of cell death are shown by PEI-PEG-Nb/pMUC1-tBid in both BT-474 (54%) and SK-BR-3 (68%) measured by luciferase activity. Introduction of tBid killer gene under the control of this promoter failed to significantly kill MCF10A cells which showed the low activity of MUC1 promoter.
Conclusion
A new horizon for treatment of cancer has been opened through cancer gene therapy. The main issues that determine the success or failure of cancer gene therapy are efficient therapeutic transgene, targeted expression and targeted delivery of the chosen transgene to cancer cells. Pro-apoptotic transgene strategy in which apoptosis could be induced in transfected cancer cells and finally could cause cell death is one of the most interesting and promising approached of cancer gene therapy. In summary, the strategy introduced in the current paper uses non-viral gene delivery systems with high specificity and low cytotoxicity to present a straightforward and effective means to accomplish active targeting. In order to target PEI polymers towards HER2-positive cells and deliver tBid killer gene to these cells, the researchers utilized anti-Her2 nanobody molecules. The evidences obtained indicate that using PEI-PEG-Nb conjugates may open a promising horizon for real applications for humans.
Acknowledgement
This work was partly supported by Tarbiat Modares University of Medical Sciences, Tehran, Iran and the Iranian Council for Stem Cell Sciences and Technologies reference [Rep 170].
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Beumer TL, Roepers-Gajadien HL, Gademan IS, et al. Apoptosis regulation in the testis: involvement of Bcl-2 family members. Mol Reprod Dev. 2000;56:353–359.
- Li H, Zhu H, Xu CJ, et al. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501.
- Luo X, Budihardjo I, Zou H, et al. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490.
- Gross A, Yin XM, Wang K, et al. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumour necrosis factor-R1/Fas death. J Biol Chem. 1999;274:1156–1163.
- Shamas-Din A, Kale J, Leber B, et al. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5:a008714.
- Gardlik R, Palffy R, Hodosy J, et al. Vectors and delivery systems in gene therapy. Med Sci Monit. 2005;11:RA110–RA121.
- Boussif O, Lezoualc'h F, Zanta MA, et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA. 1995;92:7297–7301.
- Fischer D, Bieber T, Li Y, et al. A novel non-viral vector for DNA delivery based on low molecular weight, branched polyethylenimine: effect of molecular weight on transfection efficiency and cytotoxicity. Pharm Res. 1999;16:1273–1279.
- Hong S, Leroueil P, Janus E, et al. Interaction of polycationic polymers with supported lipid bilayers and cells: nanoscale hole formation and enhanced membrane permeability. Bioconjugate Chem. 2006;17:728–734.
- Castan L, Silva CJd, Molina EF, et al. Comparative study of cytotoxicity and genotoxicity of commercial Jeffamines® and polyethylenimine in CHO-K1 cells. J Biomed Mater Res Part B: Appl Biomater. 2017.
- Merdan T, Kunath K, Petersen H, et al. PEGylation of poly(ethylene imine) affects stability of complexes with plasmid DNA under in vivo conditions in a dose-dependent manner after intravenous injection into mice. Bioconjugate Chem. 2005;16:785–792.
- Venault A, Huang YC, Lo W, et al. Tunable PEGylation of branch-type PEI/DNA polyplexes with a compromise of low cytotoxicity and high transgene expression: in vitro and in vivo gene delivery. J Mater Chem B. 2017;5:4732–4744.
- Anirudhan TS, Anila MM, Franklin S. Synthesis characterization and biological evaluation of alginate nanoparticle for the targeted delivery of curcumin. Mater Sci Eng C Mater Biol Appl. 2017;78:1125–1134.
- Pezzoli D, Tarsini P, Melone L, et al. RGD-derivatized PEI-PEG copolymers: influence of the degree of substitution on the targeting behavior. J Drug Deliv Sci Tech. 2017;37:115–122.
- Kunath K, Merdan T, Hegener O, et al. Integrin targeting using RGD-PEI conjugates for in vitro gene transfer. J Gene Med. 2003;5:588–599.
- Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013;82:775–797.
- Beghein E, Gettemans J. Nanobody technology: a versatile toolkit for microscopic imaging, protein–protein interaction analysis, and protein function exploration. Front Immunol. 2017;8.
- Jamnani FR, Rahbarizadeh F, Shokrgozar MA, et al. Targeting high affinity and epitope-distinct oligoclonal nanobodies to HER2 over-expressing tumour cells. Exp Cell Res. 2012;318:1112–1124.
- Kazhdan I, Long L, Montellano R, et al. Targeted gene therapy for breast cancer with truncated Bid. Cancer Gene Ther. 2006;13:141–149.
- Gileadi O. Methods in molecular biology. In B Kobe, M Guss, T Huber, editors. Structural proteomics: high-throughput methods. 1st ed. Humana Press. p. 426.
- Jafari Iri Sofla F, Rahbarizadeh F, Ahmadvand D, et al. Specific gene delivery mediated by poly(ethylene glycol)-grafted polyamidoamine dendrimer modified with a novel HER2-targeting nanobody. J Bioact Compat Pol. 2015;30:129–144.
- Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70–77.
- Traut RR, Bollen A, Sun TT, et al. Methyl 4-mercaptobutyrimidate as a cleavable cross-linking reagent and its application to the Escherichia coli 30S ribosome. Biochemistry. 1973;12:3266–3273.
- Ziraksaz Z, Nomani A, Ruponen M, et al. Cell-surface glycosaminoglycans inhibit intranuclear uptake but promote post-nuclear processes of polyamidoamine dendrimer-pDNA transfection. Eur J Pharm Sci. 2013;48:55–63. PubMed PMID: 23131796; eng.
- Devine PL, Birrell GW, Whitehead RH, et al. Expression of MUC1 and MUC2 mucins by human tumour cell lines. Tumour Biol. 1992;13:268–277.
- Duraisamy S, Kufe T, Ramasamy S, et al. Evolution of the human MUC1 oncoprotein. Int J Oncol. 2007;31:671–677.
- Farokhimanesh S, Rahbarizadeh F, Rasaee MJ, et al. Hybrid promoters directed tBid gene expression to breast cancer cells by transcriptional targeting. Biotechnol Progress. 2010;26:505–511.