
Abstract
Chronic inflammation in fibroblast-like synoviocytes (FLSs) induced by pro-inflammatory cytokines such as TNF-α plays a key role in the pathogenesis of rheumatoid arthritis (RA). The neurokinin-1 receptor (NK-1R) is one of the G protein–coupled receptors (GPCRs) mediating the intracellular signalling of substance P (SP). However, the possible implications of NK-1R in rheumatoid arthritis fibroblast-like synoviocytes (RA-FLSs) and the pathogenesis of RA have not yet been reported. In the current study, we report that NK-1R is expressed in FLSs. Importantly, NK-1R expression was found to be significantly increased in RA-FLSs compared to normal FLSs. Interestingly, we found that treatment with tumour necrosis factor (TNF)-α increased the expression of NK-1R at both the gene and protein levels. Treatment with the NK-1R antagonist aprepitant reduced TNF-α-induced expression of NADPH oxidase 4 (NOX-4) and generation of reactive oxygen species (ROS) in FLSs. Our results also display that blockage of NF-1R using aprepitant inhibited TNF-α-induced expression and secretion of proinflammatory cytokines, including interleukin-1β (IL-1β), IL-6, and IL-8. Consistently, aprepitant prevented TNF-α-induced expression of matrix metalloproteinases (MMPs), including matrix metalloproteinase-3 (MMP-3) and matrix metalloproteinase-13 (MMP-13). Mechanistically, our data demonstrate that treatment with aprepitant inhibited TNF-α-induced phosphorylation and degradation of inhibitor of NF-κB (IκBα). Notably, aprepitant attenuated TNF-α-induced nuclear translocation of nuclear factor κB (NF-κB) p65 and reduced luciferase activity of NF-κB in FLSs. The findings implicated a novel function of NK-1R in RA-FLSs. Blockage of NK-1R using its specific antagonist aprepitant might provide a new therapeutic strategy for RA.
Introduction
Rheumatoid arthritis (RA) is a systemic and chronic autoimmune disease with unfamiliar etiology [Citation1]. RA is pathologically characterized by synovial inflammation and hyperplasia, cartilage degradation, and damage of bones and joints [Citation2]. The synovial membrane is mainly composed of fibroblast-like synoviocytes (FLSs), which have been recognized as the principal cell type presented in aberrant pannus tissue. FLSs are important for protecting the joint by maintaining the integrity of the extracellular matrix (ECM) [Citation3]. Increasing evidence has suggested that activated rheumatoid arthritis fibroblast-like synoviocytes (RA-FLSs) play an essential role in the pathological development of arthritic joint destruction by increasing the expression and secretion of pro-inflammatory cytokines, such as tumour necrosis factor (TNF)-α, interleukin-1β (IL-1β), IL-6, and IL-8, which are involved in inducing adaptive immunity [Citation4]. Upregulation of TNF-α has been associated with exacerbation of oxidative stress in the synovial tissue, as evidenced by overproduction of reactive oxygen species (ROS) and overexpression of human NADPH oxidase 4 (NOX-4) [Citation5]. In addition, increased secretion of TNF-α causes degradation of the articular ECM, mediated by matrix metalloproteinases (MMPs), of which MMP-3 and MMP-13 have the greatest influence via the IκBα/NF-κB signalling pathway [Citation6]. Blockage of the NF-κB pathway with anti-TNF agents has been considered as an important therapeutic target of RA.
The neurokinin-1 receptor (NK-1R), an important member of three receptor subtypes for tachykinins, mediates intracellular signalling of the neuropeptide substance P (SP) [Citation7]. NK-1R is mainly located in the central nervous system (CNS). Previous studies have reported that NK-1R is found in brain regions that regulate affective behaviours and addiction [Citation8]. NK-1R activation plays a key role in regulating alcohol and drug abuse. Also, NK-1R can mediate stress- and anxiety-related effects by modulating monoaminergic neurotransmission and neuroendocrine responses [Citation9]. Interestingly, it has been recently demonstrated that NK-1R is expressed in the periphery and SP has attracted increasing attention for its physiologic actions via NK-1R in the periphery [Citation10]. Inhibition of NK-1R using its specific antagonists has been used for the treatment of addiction. Numerous NK-1R antagonists, including netupitant rolapitant, aprepitant, and casopitant, have been developed [Citation11]. However, the expression patterns of NK-1R and its biological functions in FLSs and the pathogenesis of RA have not been reported before. In the current study, we aimed to investigate the expression patterns and physiological roles of NK-1R in RA-FLSs and the TNF-α-induced inflammatory response in FLSs.
Materials and methods
FLS separation and treatment
FLSs and RA-FLSs were isolated from knee joint synovial tissues from healthy (n = 8) and RA (n = 16) donors. Experiments with human subjects were designed in accordance with the World Medical Association Declaration of Helsinki Ethical Principles for Medical Research Involving Human Subjects. Human subject experiments were approved by the ethics committee of the Beijing University of Chinese Medicine (NO. 20160032). All participants in the study signed written informed consent. Synovial biopsies were collected and cut into small pieces, followed by digestion with 0.05% trypsin and 0.5 mM ethylenediaminetetraacetic acid (EDTA) for 10 min at 37 °C under gentle shaking. Cells were collected and seeded at a density of 1.5 × 106 cells per well in 35 mm diameter cell culture dishes. Normal FLSs were stimulated with TNF-α (5, 10, 20 ng/ml) for 24 h to assess the effects of TNF-α on NK-1R expression in FLSs. Meanwhile, normal FLSs were stimulated with 10 ng/ml TNF-α in the presence or absence of aprepitant (5, 10 μM) for 24 h to detect the protective effects of aprepitant against TNF-α-induced injury.
Quantitative polymerase chain reaction (PCR) analysis
Total intracellular RNA was isolated from cells using Qiazol (Qiagen, USA) in accordance with the manufacturer’s instructions. Isolated RNA (1 µg) was used to synthesize the first-strand cDNA using an iScript cDNA synthesis kit (Bio-rad, USA). Synthesized cDNA was used for real-time PCR using an SYBR Green Supermix kit (Bio-rad, USA) on an applied real-time system (Agilent, MX3005P) to determine the expressions of target genes with the following procedures: 1 min at 95 °C, 20 s at 60 °C, 40 cycles of 5 s at 95 °C, followed by 20 s at 60 °C. Target gene expressions were calculated by normalizing to GAPDH using the 2−ΔΔct method. The following primers were used in this study:
NK-1R: forward, 5′-TCTTCTTCCTCCTGCCCTACATC-3′; reverse, 5′-AGCACCGGAAGGCATGCTTGAAGCCCA-3′;
MMP-3: forward, 5′-CCTCTATGGACCTCCCACAGAATC-3′; reverse, 5′-GGTGCTGACTGCATCGAAGGACAAA-3′;
MMP-13: forward, 5′-CTGGCCTGCTGGCTCATGCTT-3′; reverse, 5′-CCTCAGAAAGAGCAGCATCGATATG-3′;
IL-1β: forward, 5′-TTCCTGTTGTCTACACCAATGC-3′; reverse, 5′-CGGGCTTTAAGTGAGTAGGAGA-3′;
IL-6: forward, 5’-AGGGCTCTTCGGGAAATGTA-3′; reverse, 5’- TGCCCAGTGGACAGGTTTC-3′;
IL-8: forward, 5’-TTTCTGTTAAATCTGGCAACCCTAGT-3′; reverse, 5’-ATAAAGGAGAAACCAAGGCACAGT-3′;
GAPDH: forward, 5′-ACTGGCGTCTTCACCACCAT-3′; reverse, 5′-AAGGCC ATGCCAGTGAGCTT-3′.
Western blot analysis
After incubation, cells were lysed using radioimmune precipitation assay (RIPA) buffer supplemented with proteinase and phosphatase inhibitors. Samples were then centrifuged at 12,000 × g for 10 min at 4 °C to collect the supernatant. Protein concentration was measured using a commercial BCA kit (Sigma-Aldrich, USA). Cell lysates (20 µg) were subjected to 10% polyacrylamide gel and transferred to polyvinylidene fluoride (PVDF) membranes. After blocking in 5% skim milk in TBST for 2 h, membranes were sequentially loaded with primary antibodies overnight at 4 °C and HRP-conjugated secondary antibodies for 2 h at room temperature (RT). After 3 gentle washes, blots were developed using an enhanced chemiluminescence kit (Thermo Fisher Scientific, USA). The following antibodies were used in this study: anti-NK-1R (#ab61705, Abcam, USA); anti-NOX-4 (#ab133303, Abcam, USA); anti-p-IκBα (#ab133462, Abcam, USA); anti- IκBα (#ab32518, Abcam, USA); anti-NF-kB p65 (#ab16502, Abcam, USA); anti-Lamin B1 (#ab133741, Abcam, USA); anti-β-actin (#ab8226, Abcam, USA).
Measurement of intracellular reactive oxygen species (ROS)
After the indicated treatment, levels of intracellular ROS were determined using 2',7'-dichlorodihydrofluorescein diacetate (DCFH-DA) dye. FLSs were incubated with 10 ng/ml TNF-α in the presence or absence of aprepitant (5, 10 μM) for 24 h. After 3 gentle washes, FLSs were loaded with DCFH-DA (10 µM) at 37 °C and incubated for 15 min. After 3 washes, fluorescent signals were observed at 488 nm using a Zeiss Axio microscope.
Enzyme-linked immunosorbent assay (ELISA)
FLSs were incubated with 10 ng/ml TNF-α in the presence or absence of aprepitant (5, 10 μM) for 24 h. Secreted proteins of IL-1β, IL-6, IL-8, MMP-3, and MMP-13 in the supernatants were assessed using ELISA assays. The following ELISA kits were used in this study: human IL-1β ELISA Kit (#ab46052, Abcam, USA); human IL-6 ELISA Kit (#ab46027, Abcam, USA); human IL-8 ELISA Kit (#ab46032, Abcam, USA); human MMP-3 ELISA Kit (#ab100607, Abcam, USA); human MMP-13 ELISA Kit (#ab100605, Abcam, USA).
Luciferase reporter assays for NF-κB
The transcriptional activity of NF-κB was measured using a luciferase reporter gene assay. Briefly, 1 μg pNF-κB-luc or vehicle control reporter gene (Qiagen) was transfected into FLSs using the reagent Lipofectamine 2000. β-galactosidase activity was used as an internal control for transfection efficiency. At 12 h post transfection, cells were incubated with 10 ng/ml TNF-α in the presence or absence of aprepitant (5, 10 μM) for 24 h. Luciferase and β-gal activities were evaluated using a commercial luciferase reporter assay kit (Promega, USA).
Silencing of NK-1R
Gene silencing of NK-1R was performed by infecting RA-FLSs with NK-1R lentiviral short hairpin RNA (shRNA) (#sc-36069-V, Santa Cruz Biotechnology, USA). Cells were plated on 6-well plates. A mixture of complete medium with Polybrene (#sc-134220, Santa Cruz Biotechnology, USA) at a final concentration of 5 µg/ml was prepared. Culture media was removed from the plate wells and replaced with 1.5 ml polybrene/media mixture. shRNA lentiviral particles were then added to the culture to infect RA-FLSs at a multiplicity of infection (MOI) of 50.
Statistical analyses
Experimental values are presented as means ± SEM. Statistical analyses were performed using Prism software. The post hoc test S-N-K method was applied to analyze multiple comparisons following ANOVA. p Values less than .05 was considered statistically significant.
Results
NK-1R is expressed in human FLSs
Firstly, the expression of NK-1R in normal human FLSs was examined. As shown in , the results of RT-PCR indicate that NK-1R is expressed in human FLSs at the gene level. The western blot results in further confirm that NK-1R was detected in human FLSs at the protein level. Human glioblastoma cell line U251 was used as a positive control [Citation12].
Figure 1. Neurokinin 1 receptor (NK-1R) is expressed in human fibroblast-like synoviocytes (FLSs). We applied the human glioblastoma cell line U251 as a positive control. (a). RT-PCR results indicate that NK-1R is expressed in human FLSs at the gene level; (b). Western blot results indicate that NK-1R is expressed in human FLSs at the protein level.

Expression of NK-1R is increased in RA-FLSs and TNF-α-treated normal FLSs
The differences in expression of NK-1R between normal human FLSs and human RA-FLSs were compared. The results of real-time PCR in and western blot analysis in indicate that NK-1R expression was significantly increased in RA-FLSs as compared to that of FLSs at both the mRNA and protein levels. Overproduction of TNF-α has been associated with the pathological development of RA. Therefore, the expression level of NK-1R in response to TNF-α treatment in FLSs was investigated. FLSs were treated with TNF-α at the concentrations of 5, 10, 20 ng/ml. The results of real-time PCR in and western blot analysis in indicate that expression of NK-1R was increased by treatment with TNF-α in a concentration-dependent manner.
Figure 2. Expression of neurokinin 1 receptor (NK-1R) is increased in rheumatoid arthritis (RA)-FLSs as compared to normal FLSs. (a). Real-time PCR analysis of NK-1R; (b). Western blot analysis of NK-1R (*, p < .01 vs. vehicle group).
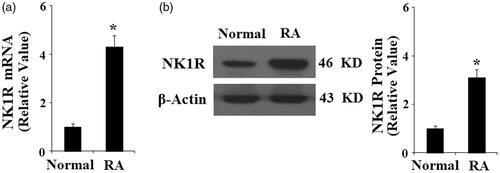
Figure 3. Expression of neurokinin 1 receptor (NK-1R) is increased in normal FLSs in response to TNF-α treatment (5, 10, 20 ng/ml) in a concentration-dependent manner. (a) Expression of NK-1R at the gene level was determined by real-time PCR analysis; (b). Expression of NK-1R at the protein level was determined by Western blot analysis (*, #, $, p < .01 vs. previous column group).
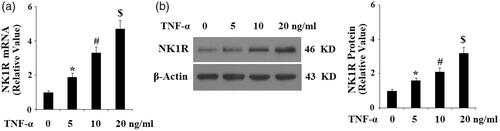
Antagonism of NK-1R with aprepitant ameliorates TNF-α-induced oxidative stress in FLSs
These results suggest that NK-1R might play a role in the pathogenesis of RA. Therefore, we investigated whether the NK-1R antagonist aprepitant has a protective effect against TNF-α-induced insult in FLSs. The results in display that treatment with aprepitant significantly reduced TNF-α (10 ng/ml)-induced production of ROS, as determined by DCFH-DA assay, in a dose-dependent manner (5, 10 μM). Human NADPH oxidase 4 (NOX-4) is the main source of ROS [Citation13]. Importantly, we found that the presence of aprepitant significantly inhibited the expression of NK-1R (). These results suggest that blockage of NK-1R using aprepitant prevents TNF-α-induced oxidative stress in FLSs.
Figure 4. The neurokinin-1 receptor (NK-1R) antagonist aprepitant ameliorates TNF-α-induced reactive oxygen species (ROS) production and NOX-4 expression in FLSs. FLSs were incubated with 10 ng/ml TNF-α in the presence or absence of aprepitant (5, 10 μM) for 24 h. (a). ROS generation was determined by DCFH-DA assay; Scale bars, 100 μm; (b). NOX-4 expression was determined by Western blot analysis (*, #, $, p < .01 vs. previous column group).
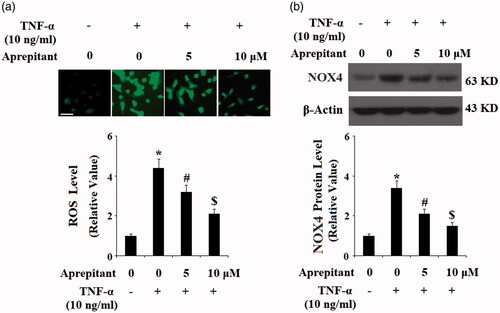
Antagonism of NK-1R with aprepitant reduces expression and secretion of pro-inflammatory cytokines in TNF-α-treated FLSs
We then assessed the effects of aprepitant in TNF-α-induced expression of pro-inflammatory cytokines, including IL-1β, IL-6, and IL-8. The results of real-time PCR in indicate that treatment with aprepitant significantly reduced TNF-α-induced expression of IL-1β, IL-6, and IL-8 at the mRNA level. Consistently, the ELISA results in indicate that treatment with aprepitant inhibited TNF-α-induced secretion of these cytokines.
Figure 5. The neurokinin-1 receptor (NK-1R) antagonist aprepitant inhibits TNF-α (10 ng/ml)-induced expression of IL-1β, IL-6, IL-8 in FLSs. FLSs were incubated with 10 ng/ml TNF-α in the presence or absence of aprepitant (5, 10 μM) for 24 h. (a). mRNA expression of IL-1β, IL-6, and IL-8 determined by real-time PCR; (b). Secretion of IL-1β, IL-6 and IL-8 determined by ELISA (*, #, $, p<.01 vs. previous column group).
Antagonism of NK-1R with aprepitant reduces expression and secretion of MMP-3 and MMP-13 in TNF-α-treated FLSs
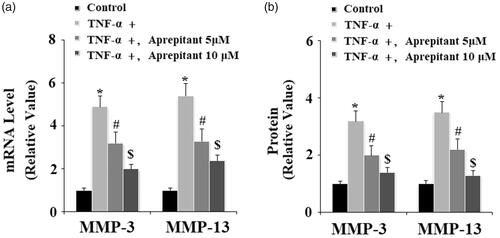
Upregulation of MMP-3 and MMP-13 in FLSs has been considered as an important biomarker of the pathological development of RA [Citation14]. Here, the effects of aprepitant on TNF-α-induced expression of MMP-3 and MMP-13 were studied. The results of real-time PCR in and western blot analysis in indicate that the presence of aprepitant significantly decreased TNF-α-induced expression of MMP-3 and MMP-13 at both the gene and protein levels.
Figure 6. The neurokinin-1 receptor (NK-1R) antagonist aprepitant inhibits TNF-α (10 ng/ml)-induced expression of MMP-3 and MMP-13 in FLSs. FLSs were incubated with 10 ng/ml TNF-α in the presence or absence of aprepitant (5, 10 μM) for 24 h. (a). mRNA expression of MMP-3 and MMP-13 determined by real-time PCR; (b). Protein expression of MMP-3 and MMP-13 determined by ELISA (*, #, $, p < .01 vs. previous column group).
Antagonism of NK-1R with aprepitant inhibits TNF-α-induced activation of IκBα/NF-κB signalling
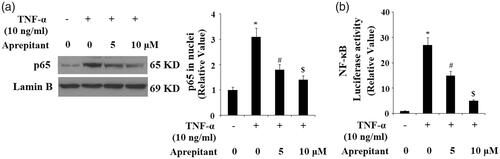
We further studied the signalling pathway of aprepitant and NK-1R by detecting levels of phosphorylated and total IκBα by western blot analysis. The results indicate that TNF-α treatment significantly increased the level of phosphorylated IκBα and induced the degradation of total IκBα. Importantly, these effects were ameliorated by treatment with aprepitant (. Degradation of IκBα leads to nuclear translocation of NF-κB p65, which participates in the transcription of pro-inflammatory factors. Nuclear fractions were used to determine the expression of p65 in nuclei with lamin B as a positive control. As expected, the results in indicate that treatment with aprepitant significantly ameliorated TNF-α-induced p65 expression in nuclei. Importantly, the results of luciferase reporter assay in indicate that NF-κB activity induced by TNF-α was reduced by the presence of aprepitant.
Figure 7. The neurokinin-1 receptor (NK-1R) antagonist aprepitant inhibits TNF-α (10 ng/ml)-induced expression of p-IκBα and IκBα degradation in FLSs. FLSs were incubated with 10 ng/ml TNF-α in the presence or absence of aprepitant (5, 10 μM) for 6 h. Western blot analysis showing amounts of p-IκBα and IκBα in FLSs (*, #, $, p<.01 vs. previous column group).
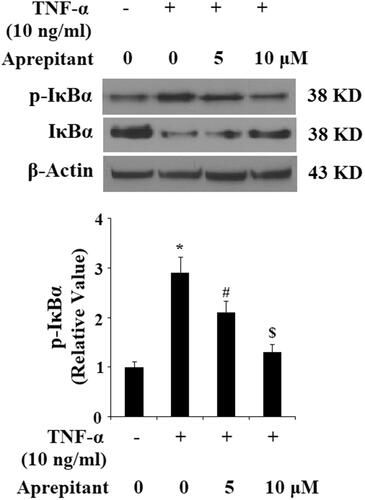
Figure 8. The neurokinin-1 receptor (NK-1R) antagonist aprepitant inhibits TNF-α (10 ng/ml)-induced nuclear translocation of p65 and luciferase activity of NF-κB. (a) Nuclear levels of p65 was determined by Western blot analysis with lamin B as a positive control; (b) Luciferase reporter assay results reveal that NF-κB activity was attenuated by treatment with aprepitant (*, #, $, p < .01 vs. previous column group).
Knockdown of NK-1R in RA-FLSs inhibits expression of MMP-3, MMP-13 and activation of NF-κB
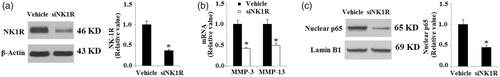
To further confirm the function of NK-1R in the inflammatory response in RA-FLSs, expression of NK-1R was knocked down by infection with NK-1R lentiviral shRNA in RA-FLSs. Successful knockdown of NK-1R is shown in . We found that silencing of NK-1R reduced expression of MMP-3 and MMP-13 in RA-FLSs (). Notably, we found that silencing of NK-1R prevented nuclear translocation of NF-κB p65, suggesting that NK-1R knockdown ameliorates the inflammatory response in RA-FLSs.
Figure 9. Silencing of NK-1R ameliorates the inflammatory response in RA-FLSs. RA-FLSs were infected with NK-1R lentiviral shRNA for 24 h. (a). Western blot analysis revealed successful knockdown of NK-1R; (b). Expression of MMP-3 and MMP-13 at the gene level determined by real-time PCR analysis; (c). Nuclear translocation of NK-1R determined by Western blot analysis (*, p < .01 vs. vehicle control).
Discussion
The main pathological characteristics of RA are defined as chronic synovial inflammation associated with high levels of proinflammatory cytokines, and injury to bone tissue and cartilage. A variety of studies have shown that FLSs play a pivotal role in the occurrence and development of RA [Citation15]. However, the underlying mechanisms have not been elucidated. In the current study, we investigated expression patterns and biological functions in RA-FLSs. Firstly, we report for the first time to our knowledge, that NK-1R is expressed in FLSs. Notably, we found that expression of NK-1R is upregulated in RA-FLSs. NK-1R is increased in normal FLSs in response to TNF-α treatment. NK-1R, a seven-transmembrane domain G-protein coupled receptor, acts as a mediator of the inflammatory effects of substance P (SP). Abnormal expression of NK-1R has been linked with the pathophysiological progression of a variety of diseases [Citation16]. Activation of NK-1R signalling can augment inflammatory processes by promoting immune cell infiltration and the release of cytokines [Citation17]. Our findings implicate that upregulation of NK-1R might be linked to the inflammatory process of FLSs and the pathological progression of RA. Aprepitant is the first NK-1R antagonist licensed to block chemotherapy-induced nausea and vomiting [Citation18]. Studies have revealed that NK-1R antagonists are efficacious, safe, and well tolerated for clinical use in most of the population [Citation19]. The activity of NK-1R is blocked by its specific antagonist aprepitant. Furthermore, we also found that treatment with aprepitant significantly suppressed TNF-α-induced expression of IL-1β, IL-6, and IL-8 as well as MMP-3 and MMP-13, suggesting that aprepitant may prevent augmentation of inflammatory reactions induced by TNF-α. Mechanistically, treatment with aprepitant inhibited activation of the IκBα/NF-κB signalling pathway, which acts as a central regulator governing inflammatory signalling.
Excessive production of proinflammatory mediators such as IL-1β, IL-6, IL-8, and TNF-α as well as the increased generation of ROS play an important role in affecting the progression and severity of RA. Blockage of oxidative stress and inflammatory responses induced by pro-inflammatory factors in synoviocytes has been considered as a potential therapeutic strategy against RA. For example, salicin has been shown to display anti-oxidant and anti-inflammatory capacities through hindering NF-κB signalling and activating Nrf2 and HO-1 in RA-FLSs [Citation20]. Another study reported that administration of the natural product paeonol could attenuate IL-1β-induced synovial inflammation in FLS-RAs and progression of RA in mice through modulating the production of proinflammatory cytokines and activation of the TLR4/NF-κB signalling pathway [Citation21]. Imperatorin has been reported to inhibit cell proliferation and induce apoptosis in RA-FLSs, which may be associated with mitochondrial/caspase-mediated signalling pathways [Citation22]. TNF-α has been identified as a predominant inflammatory regulator in the pathogenesis of RA. The expression of TNF-α is greatly increased in synovial fluid in RA [Citation23]. TNF-α stimulation in FLSs can induce a prolonged and sustained inflammatory response, leading to persistent synovial inflammation in RA [Citation24]. Thus, disrupting the TNF-α signalling cascade in RA FLSs has been considered as an important strategy for the treatment of RA. Indeed, TNF-α inhibitors have made great advances in the management and therapy of RA [Citation25]. In this study, we report that blockage of NK-1R using aprepitant ameliorates the TNF-α-induced inflammatory response in FLSs. Consistently, the anti-inflammatory effects of aprepitant have been reported in previous investigations. For example, antagonism of NK-1R using aprepitant has been shown to attenuate the B. burgdorferi-associated neuroinflammatory response by preventing astrocyte activation [Citation26]. NOX-4 is involved in the release of superoxide by FLSs in response to TNF-α treatment [Citation27]. Interestingly, we found that aprepitant treatment reduced NOX-4 expression and production of ROS, suggesting that antagonism of NK-1R may be able to mitigate oxidative stress in RA. The presence of increased expression of cytokines and upregulated expression of MMP-3 and MMP-13 in RA-FLSs leads to collagen degradation and joint inflammation in RA [Citation6]. Our findings show that aprepitant treatment reduced the expression of MMP-3 and MMP-13, implicating that aprepitant might possess a protective effect against articular cartilage disruption in RA. TNF-α initiates the expression of pro-inflammatory genes by activating the NF-κB signalling pathway. NF-κB is a central regulator of the inflammatory response in RA-FLSs. The inhibitory effects of aprepitant in NF-κB activation suggests that aprepitant may have a wide range of anti-inflammatory capacities in FLSs.
In summary, we report a novel biological function of NK-1R in regulating the inflammatory response in RA-FLSs. Blockage of NK-1R with its specific antagonist aprepitant could mitigate the release of inflammatory factors in RA-FLSs.
Disclosure statement
None of the authors of this article have any conflicts of interest that need to be disclosed.
Additional information
Funding
Related Research Data
References
- McGonagle D, Watad A, Savic S. Mechanistic immunological based classification of rheumatoid arthritis. Autoimmun Rev. 2018;17:1115–1123.
- Zhang A, Lee YC. Mechanisms for joint pain in rheumatoid arthritis (RA): from cytokines to central sensitization. Curr Osteoporos Rep. 2018;16:603–610.
- Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9:24–33.
- Bustamante MF, Garcia-Carbonell R, Whisenant KD, et al. Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthritis Res Ther. 2017;19:110.
- Phull AR, Nasir B, Haq IU, et al. Oxidative stress, consequences and ROS mediated cellular signaling in rheumatoid arthritis. Chem Biol Interact. 2018;281:121–136.
- Araki Y, Mimura T. Matrix metalloproteinase gene activation resulting from disordred epigenetic mechanisms in rheumatoid arthritis. IJMS. 2017;18:905.
- Schank JR, Heilig M. Substance P and the neurokinin-1 receptor: the New CRF. Int Rev Neurobiol. 2017;136:151–175.
- Yip J, Chahl LA. Localization of tachykinin receptors and Fos-like immunoreactivity induced by substance P in guinea-pig brain. Clin Exp Pharmacol Physiol. 2000;27:943–946.
- Ebner K, Muigg P, Singewald G, et al. Substance P in stress and anxiety: NK-1 receptor antagonism interacts with key brain areas of the stress circuitry. Ann N Y Acad Sci. 2008;1144:61–73.
- Gautam M, Prasoon P, Kumar R, et al. Role of neurokinin type 1 receptor in nociception at the periphery and the spinal level in the rat. Spinal Cord. 2016;54:172–182.
- Bošnjak SM, Gralla RJ, Schwartzberg L. Prevention of chemotherapy-induced nausea: the role of neurokinin-1 (NK1) receptor antagonists. Support Care Cancer. 2017;25:1661–1671.
- Mou L, Kang Y, Zhou Y, et al. Neurokinin-1 receptor directly mediates glioma cell migration by up-regulation of matrix metalloproteinase-2 (MMP-2) and membrane type 1-matrix metalloproteinase (MT1-MMP). J Biol Chem. 2013;288:306–318.
- Drevet S, Gavazzi G, Grange L, et al. Reactive oxygen species and NADPH oxidase 4 involvement in osteoarthritis. Exp Gerontol. 2018;111:107–117.
- Hayashi N, Nishimura K, Kumagai S. New biomarkers for rheumatoid arthritis. Rinsho Byori. 2008;56:297–308.
- Audo R, Hegglin A, Severac D, et al. Identifcation of genes regulating TRAIL-induced apoptosis in rheumatoid arthritis fbroblasts-like synoviocytes. Genes Immun. 2015;16:462–469.
- Garcia-Recio S, Gascón P. Biological and pharmacological aspects of the NK1-receptor. Biomed Res Int. 2015;2015:495704.
- Johnson MB, Young AD, Marriott I. The therapeutic potential of targeting substance P/NK-1R interactions in inflammatory CNS disorders. Front Cell Neurosci. 2017;10:296.
- Patel L, Lindley C. Aprepitant-a novel NK1-receptor antagonist. Expert Opin Pharmacother. 2003;4:2279–2296.
- Quartara L, Altamura M. Tachykinin receptors antagonists: from research to clinic. Curr Drug Targets. 2006;7: 975–992.
- Zhai KF, Duan H, Khan GJ, et al. Salicin from alangium chinense ameliorates rheumatoid arthritis by modulating the Nrf2-HO-1-ROS pathways. J Agric Food Chem. 2018;66:6073–6082.
- Zhai KF, Duan H, Luo L, et al. Protective effects of paeonol on inflammatory response in IL-1β-induced human fibroblast-like synoviocytes and rheumatoid arthritis progression via modulating NF-κB pathway. Inflammopharmacology. 2017 Aug 10. doi: 10.1007/s10787-017-0385-5.
- Zhai KF, Duan H, Chen Y, et al. Apoptosis effects of imperatorin on synoviocytes in rheumatoid arthritis through mitochondrial/caspase-mediated pathways. Food Funct. 2018;9:2070–2079.
- Gu X, Gu B, Lv X, et al. 1, 25-dihydroxy-vitamin D3 with tumor necrosis factor-alpha protects against rheumatoid arthritis by promoting p53 acetylation-mediated apoptosis via Sirt1 in synoviocytes. Cell Death Dis. 2016;7:e2423.
- Lee A, Qiao Y, Grigoriev G, et al. Tumor necrosis factor alpha induces sustained signaling and a prolonged and unremitting inflammatory response in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol. 2013;65:928–938.
- Lim H, Lee SH, Lee HT, et al. Structural biology of the TNFα antagonists used in the treatment of rheumatoid arthritis. Int J Mol Sci. 2018 Mar 7;19. pii: E768.
- Martinez AN, Burmeister AR, Ramesh G, et al. Aprepitant limits in vivo neuroinflammatory responses in a rhesus model of Lyme neuroborreliosis. J Neuroinflammation. 2017;14:37.
- Chenevier-Gobeaux C, Lemarechal H, Bonnefont-Rousselot D, et al. Superoxide production and NADPH oxidase expression in human rheumatoid synovial cells: regulation by interleukin-1beta and tumour necrosis factor-alpha. Inflamm Res. 2006;55:483–490.