
Abstract
Hepatitis B virus X protein (HBx), encoded by the hepatitis B virus (HBV) genome, plays a pivotal in mediating pathogenicity in liver diseases. However, the underlying mechanisms are still needed to be elucidated. Receptor-interacting protein 1 (RIP1) has been identified as a serine/threonine kinase with various biological functions in cell fate, proliferation, and death. In the current study, we found that overexpression of HBx in LO2 human normal hepatocytes increased the expression of RIP1. Importantly, our results indicate that blockage of RIP1 using its specific antagonist necrostatin-1 (Nec-1) ameliorated HBx-induced oxidative stress by mitigating the production of ROS and Nox-4 expression. Also, the presence of Nec-1 improved HBx-induced mitochondrial dysfunction by increasing MMP. Importantly, we found that Nec-1 could inhibit the production of pro-inflammatory cytokines IL-6, IL-8, and CXCL2 as well as the secretion of HMGB1. Mechanistically, we found that Nec-1 treatment suppressed the activation of the JNK/AP-1 and NF-κB signalling pathways. Our findings implicated a novel biological function of RIP1 in HBx-induced cytotoxicity and inflammation in human normal hepatocytes. Antagonism of RIP1 might be a potential therapeutic approach for the treatment of HBV-associated liver diseases.
Keywords:
Introduction
Hepatitis B virus (HBV) infection is recognized as one of the major causes of chronic liver diseases, which have become a global health problem and affect millions of people worldwide [Citation1]. HBV X protein (HBx), a protein containing 154 amino acids, is encoded by the smallest open reading frame of the DNA genome of HBV [Citation2]. Studies have shown that HBx acts as a regulatory protein with pleiotropic effects. HBx plays an important role in enhancing HBV replication and causing various damages to hepatocytes [Citation3]. HBx has recently been reported to induce autophagosome formation by increasing the production of reactive oxygen species (ROS) and activation of JNK [Citation4]. Importantly, HBx causes apoptosis of hepatocytes through promoting the translocation of Bax from the cytoplasm to mitochondria, reducing mitochondrial membrane potential (MMP), and stimulating the release of cytochrome C [Citation5]. Additionally, HBx treatment causes excessive production of several pro-inflammatory cytokines, such as interleukin-6 (IL-6), IL-8, chemokine (C-X-C motif) ligand 2 (CXCL2), and transforming growth factor-β (TGF-β), which have been considered as essential contributors to chronic liver inflammation and fibrosis [Citation6]. Importantly, HBx could transactivate multiple intracellular signalling pathways including AP1 and NF-κB [Citation7], which act as key regulators of inflammatory reactions in various tissue and cell types. However, the underlying molecular mechanisms are still needed to be elucidated.
Receptor-interacting protein 1 (RIP1), a serine/threonine kinase, is an important regulator of necroptosis execution by modulating the necrosome formation [Citation8]. RIP1 is well unknown to mediate the pro-inflammatory cytokine tumour necrosis factor-α (TNF-α)-induced inflammation and cell death through activating the transcriptional factor NF-κB [Citation9]. Also, RIP1 is also able to mediate TNF-α-induced activation of several intracellular kinases, inducing the mitogen-activated protein kinase (MAPK) JNK [Citation10]. The contribution and the involvement of RIP1-mediated necroptosis in inflammatory diseases have been extensively studied. Interestingly, RIP1 participates in regulating IL-1β maturation via activation of the NLRP3 inflammasome [Citation11]. Necrostatin-1 (Nec-1) is originally developed as an inhibitor of RIP1 and necroptosis. Pharmacological activities have been studied in a diversity of disease models [Citation12]. Nec-1 exerts a protective effect against N-methyl-d-aspartic acid receptor (NMDA)-induced excitotoxicity in rat's cultured cortical neurons [Citation13]. However, the physiological roles of RIP1 and the pharmacological functions of Nec-1 in HBV X protein-mediated toxicity in hepatocytes have not been reported before.
Materials and methods
Cell culture and transfection
LO2 human hepatocytes were purchased from ATCC, Manassas, VA. Cells were cultured in DMEM supplemented with 10% FBS, 1% penicillin–streptomycin. A HBx genome sequence [Citation14] was subcloned into the pCDNA3.1 vector (Invitrogen, Carlsbad, CA). About 5 × 105 LO2 hepatocytes were seeded into each well on a 6-well cell culture plate and incubated in normal medium for 12 h. About 2 μg pCDNA3. 1-HBx plasmid or empty control DNA was transfected into cells using lipofectamine 2000 (Invitrogen, Carlsbad, CA) as described previously. Successful overexpression of HBx was determined by western blot analysis.
Western blot analysis
Proteins were extracted from LO2 hepatocytes using cell lysis buffer (Cell Signalling Technologies, Danvers, MA) containing phosphatase and protease inhibitors cocktail (Roche, Indianapolis, IN). The protein concentration was examined using a pierce BCA protein assay kit (Thermo Fisher Scientific Technologies, Waltham, MA). Samples were run on 10% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. Non-specific sites on the membranes were blocked with 5% fat-skim milk in TBST for 2 h at room temperature (RT). Then the PVDF membranes were sequentially probed with primary antibodies for 3 h and HRP-linked secondary antibodies for 1 h at RT. Specific bands were detected with an ECL western blotting substrate kit (Thermo Fisher Scientific Technologies, Waltham, MA).
Real-time PCR analysis
Total RNA was extracted from LO2 cells using Qiazol (Qiagen, Valencia, CA). About 2 μg mRNA was used to make cDNA with iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) in a reverse transcription PCR (RT-PCR). About 2 μl RT-PCR product was used for real-time PCR using SYBR green qPCR master mix (Thermo Fisher Scientific Technologies, Waltham, MA) with the following procedure: 95 °C for 1 min, 40 cycles of 95 °C for 5 s, 60 °C for 30 s on an ABI 7500 real-time PCR system. Expressions of target gene were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Reactive oxygen species (ROS) measurement
Intracellular levels of ROS in LO2 hepatocytes were assessed using 2',7'-dichlorodihydrofluorescein diacetate (DCFH-DA) (Sigma-Aldrich, St. Louis, MO). LO2 normal hepatocytes were transfected with a HBx-encoding plasmid. Twenty-four hour later, cells were treated with Nec-1 at the concentration of 50 and 100 μM for another 24 h. Then cells were probed with 5 μM DCFH-DA in phenol red free medium for 30 min. Fluorescent signals were visualized in the IBE2000 inverted fluorescence microscope (Zeiss, Oberkochen, Germany).
Mitochondrial membrane potential (MMP) determination
Levels of MMP in LO2 hepatocytes were evaluated using tetramethylrhodamine methyl ester (TMRM) (Invitrogen, Carlsbad, CA). LO2 normal hepatocytes were transfected with HBx-encoding plasmid. Twenty-four hour later, cells were treated with Nec-1 at the concentration of 50 and 100 μM for another 24 h. Then cells were probed with 20 nmol/L TMRM in phenol red free medium for 30 min. Fluorescent signals were visualized in the IBE2000 inverted fluorescence microscope (Zeiss, Oberkochen, Germany).
Enzyme-linked immunosorbent assay (ELISA) analysis
LO2 normal hepatocytes were transfected with HBx-encoding plasmid. 24 h later, cells were treated with Nec-1 at the concentration of 50 and 100 μM for another 24 h. Secretions of IL-6, IL-8, CXCL2, and HMGB1 from LO2 normal hepatocytes to the cell culture medium were measured using ELISA kits (R&D Systems, Minneapolis, MN) in accordance with the manufacturer’s protocols.
Luciferase reporter gene assay
One microgram reporter plasmid (either pNF-κB-luc, AP-1-luc or negative control) (Qiagen, Valencia, CA) was transfected into LO2 normal hepatocytes with Lipofectamine 2000 (Invitrogen, Carlsbad, CA). β-Galactosidase activity was used to control transfection efficiency. Activities of luciferase and β-gal were detected using a commercial kit from Promega, Madison, WI. Luciferase activity was normalized to β-galactosidase activity and used to index transcriptional activities of AP-1 or NF-κB.
Statistical analysis
Experimental results are expressed as the means ± SE. Statistical comparisons among different groups were made using ANOVA. p < .05 was considered statistically significant.
Results
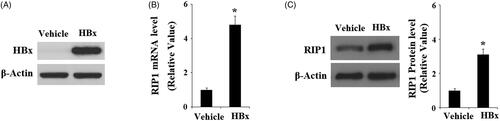
To determine the effects of HBx on the expression of RIP1, we analyzed the expression of RIP1 in LO2 hepatocytes upon enforced expression of HBx. Successful overexpression of RIP1 was verified by western blot analysis, shown in . Interestingly, both real-time PCR () and western blot analysis () revealed that the expression of RIP1 was significantly increased in response to HBx overexpression at both the mRNA levels and protein levels. These results suggest that RIP1 might play a role in mediating the cellular toxicity of HBx.
Figure 1. Induction of RIP1 by HBx in human LO2 normal hepatocytes. Human LO2 normal hepatocytes were transfected with HBx-encoding plasmid for 48 h. (A) Successful transfection of HBx was confirmed by western blot analysis. (B) Real-time PCR analysis revealed that overexpression of HBx increased the expression of RIP1 at the mRNA level. (C) Western blot analysis revealed that overexpression of HBx increased the expression of RIP1 at the protein level (*, p < .01 versus the vehicle group).
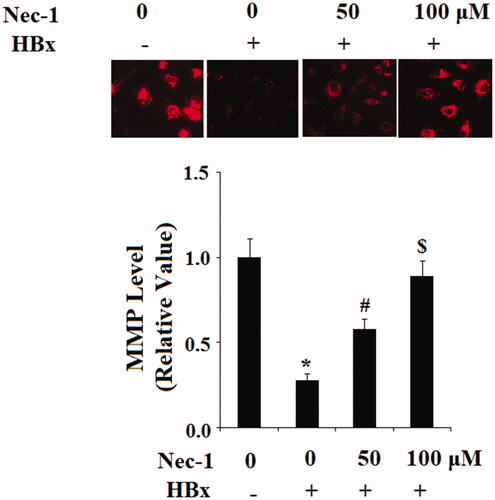
To study the possible involvement of RIP1 on HBx-induced oxidative stress in LO2 hepatocytes, cells were transfected with the HBx-encoding plasmid. Twenty-four hour later, cells were treated with Nec-1 at the concentrations of 50 and 100 μM for 24 h. DCFH-DA staining results indicated that HBx overexpression markedly increased the production of intracellular ROS, which was mitigated by treatment with Nec-1 in a dose-dependent manner (). NADPH oxidase 4 (Nox-4) plays an important role in mediating the production of mitochondrial ROS. Here, our results indicate that HBx overexpression significantly increased the expression of Nox-4. However, the presence of Nec-1 decreased the expression of Nox-4 in a dose-dependent manner (). Mitochondrial function was assessed by TMRM staining. Results in indicated that HBx overexpression significantly reduced MMP levels in LO2 hepatocytes. As expected, the presence of Nec-1 reserved the reduction of MMP induced by HBx overexpression.
Figure 2. The RIP1 inhibitor necrostatin-1 (Nec-1) ameliorated HBx-induced oxidative stress in human LO2 normal hepatocytes. Human LO2 normal hepatocytes were transfected with HBx-encoding plasmid. Twenty-four hour later, cells were treated with Nec-1 at the concentration of 50 and 100 μM for 24 h. (A) Reactive oxygen species (ROS) was determined by the DCFH-DA assay. (B) Expression of Nox-4 was determined by western blot analysis (*, #, $, p < .01 versus the previous column group).
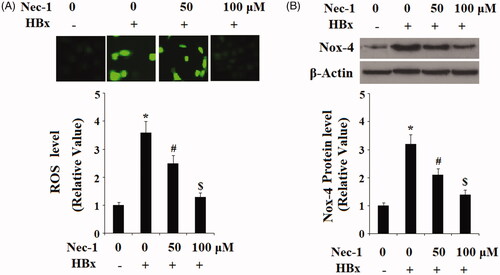
Figure 3. The RIP1 inhibitor necrostatin-1 (Nec-1) ameliorated HBx-induced reduction of mitochondrial membrane potential (MMP) in human LO2 normal hepatocytes. LO2 normal hepatocytes were transfected with HBx-encoding plasmid. Twenty-four hour later, cells were treated with Nec-1 at the concentration of 50 and 100 μM for another 24 h. Intracellular level of mitochondrial membrane potential (MMP) was determined by TMRM staining (*, #, $, p < .01 versus the previous column group).
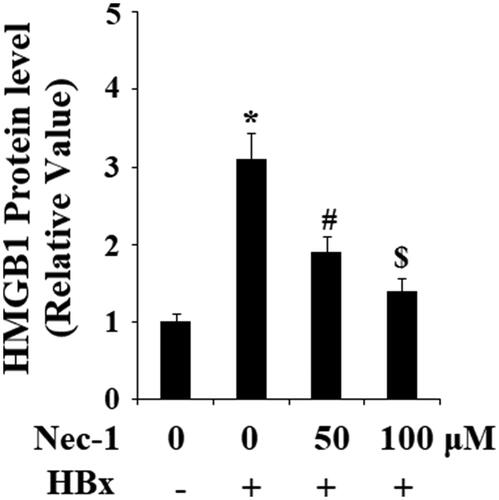
HBx has been reported to induce the expression and secretion of various pro-inflammatory cytokines. Here, the effects of Nec-1 on the expressions of IL-6, IL-8, and CXCL2 were assessed. Real-time PCR results in showed that HBx overexpression remarkably upregulated the expression of IL-6, IL-8, and CXCL2. As expected, Nec-1 obviously inhibited the expression of IL-6, IL-8, and CXCL2 in a concentration-dependent manner. Secretion of IL-6, IL-8, and CXCL2 was then examined using ELISA kits. As expected, results in indicated that HBx overexpression-induced the secretion of IL-6, IL-8, and CXCL2 was significantly ameliorated by Nec-1 in a dose-dependent manner. High-mobility group box 1 protein (HMGB1) is a well-known conserved chromosomal protein, which can be released from damaged cells. To assess the effects of Nec-1 on HBx-induced cell damage, secretion of HMGB1 from LO2 hepatocytes was examined by an ELISA kit. Results in indicated that HBx overexpression significantly promoted the secretion of HMGB1, which was prevented by Nec-1 in a dose-dependent manner.
Figure 4. The RIP1 inhibitor necrostatin-1 (Nec-1) reduced HBx-induced expressions and secretions of IL-6, IL-8, and CXCL2. LO2 normal hepatocytes were transfected with HBx-encoding plasmid. 24 h later, cells were treated with Nec-1 at the concentration of 50 and 100 μM for another 24 h. (A) Expression of IL-6, IL-8, and CXCL2 at the mRNA levels was determined by real-time PCR analysis. (B) Expression of IL-6, IL-8, and CXCL2 at the protein levels was determined by ELISA assay (*, #, $, p < .01 versus the previous column group).
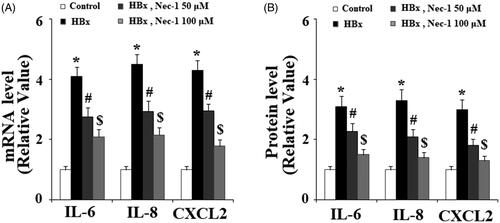
Figure 5. The RIP1 inhibitor necrostatin-1 (Nec-1) reduced HBx-induced secretion of high mobility group box 1 (HMGB1). LO2 normal hepatocytes were transfected with HBx-encoding plasmid. Twenty-four hour later, cells were treated with Nec-1 at the concentration of 50 and 100 μM for another 24 h. Secretion of HMGB1 was determined by ELISA (*, #, $, p < .01 versus the previous column group).
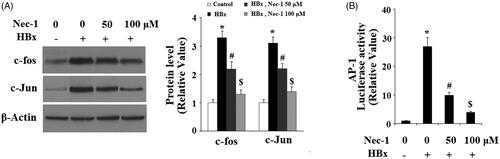
ROS trigger activation of several signalling pathways including c-Jun N-terminal kinase (JNK). The activation of JNK is reported to participate in mediating HBx-induced cytotoxicity [Citation15]. Here, our results indicate that HBx overexpression significantly increased the phosphorylation of JNK (), which was attenuated by treatment with Nec-1 in a dose-dependent manner. Phosphorylation of JNK initiates the activation of transcriptional factor AP-1, triggering the expression of downstream genes. Here, we found that HBx overexpression significantly increased the expression of c-fos and c-Jun (), which are the two major components of AP-1. However, this process was prevented by the treatment with Nec-1. To test the transcriptional activity of AP-1, luciferease activity of the AP-1 reporter gene was assayed. Notably, it was shown that Nec-1 treatment significantly inhibited HBx overexpression-induced increase in the luciferase activity of AP-1, suggesting the essential roles of the JNK/AP-1 signalling pathway in the inhibitory effects of Nec-1 in cytotoxicity of HBx.
Figure 6. The RIP1 inhibitor necrostatin-1 (Nec-1) inhibited HBx-induced phosphorylation of JNK. LO2 normal hepatocytes were transfected with HBx-encoding plasmid. Twenty-four hour later, cells were treated with Nec-1 at the concentration of 50 and 100 μM for another 2 h. Phosphorylated and total levels of JNK were determined by western blot analysis(*, #, $, p < .01 versus the previous column group).
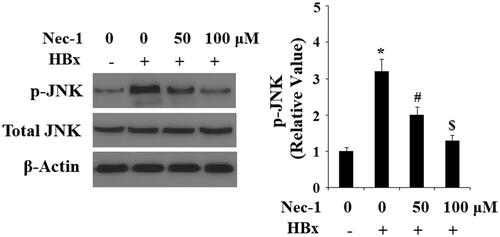
Figure 7. The RIP1 inhibitor necrostatin-1 (Nec-1) prevented HBx-induced activation of AP-1. LO2 normal hepatocytes were transfected with HBx-encoding plasmid. Twenty-four hour later, cells were treated with Nec-1 at the concentration of 50 and 100 μM for another 24 h. (A) Expression of the AP-1 components c-fos and c-Jun was determined by western blot analysis. (B) Luciferase activity of AP-1 promoter (*, #, $, p < .01 versus the previous column group).
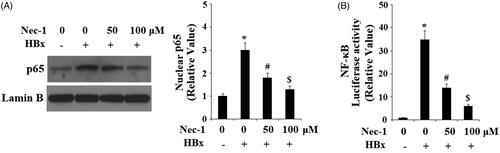
The activation of NF-κB plays a central role in inflammation reaction mediated by nuclear translocation of NF-κB p65. Here, our findings indicated that HBx overexpression significantly increased the nuclear translocation of p65. However, the presence of Nec-1 prevented this process (). Consistently, luciferase activity assay reported that Nec-1 mitigated HBx overexpression-induced increase in the luciferase activity of the NF-κB promoter ().
Figure 8. The RIP1 inhibitor necrostatin-1 (Nec-1) prevented HBx-induced activation of NF-κB. LO2 normal hepatocytes were transfected with HBx-encoding plasmid. Twenty-four hour later, cells were treated with Nec-1 at the concentration of 50 and 100 μM for another 24 h. (A) Nuclear translocation of NF-κB p65 was determined by western blot analysis with Lamin B as a positive control. (B) Luciferase activity of NF-κB (*, #, $, p < .01 versus the previous column group).
Discussion
Chronic infection with HBV has been associated with several liver diseases including hepatitis, cirrhosis, and hepatocellular carcinoma (HCC) [Citation16]. HBx, a 16.5-kDa regulatory protein, is one of the main components of HBV. HBx possesses multifunctional activities including enhancing HBV replication. Although the biological functions of HBx in hepatocytes have been intensively studied in the past decades, the immune mechanisms of liver damage are still needed to be elucidated. In the current study, our findings demonstrate, for the first time, that HBx induced the expression of RIP1 in LO2 hepatocytes. Importantly, we found that treatment with the RIP1-specific antagonist could ameliorate HBx-induced cytotoxicity and inflammation in LO2 hepatocytes. HBx is often reported to locate in mitochondria and induces mitochondrial dysfunction by reducing MMP, which has been associated with hepatocyte death [Citation17] and oxidative liver injury [Citation18]. The production of ROS has also been linked with the physiological function of RIP1. N-Acetyl-l-cysteine (NAC), a well-known ROS scavenger, has been shown to abolish RIP1-mediated necroptosis [Citation19]. Here, our results indicate that Nec-1 treatment ameliorated HBx-induced production of ROS, Nox-4 expression, and reduction of MMP. Pro-inflammatory cytokines are important for immune-associated liver damage in response to HBV infection. For example, HBx is able to induce the production of IL-1β, IL-6, IL-8, TNF-α, and CXCL2 through promoting the expression of IL-32, a newly identified cytokine [Citation20]. In the current study, we found that Nec-1 reduced HBx overexpression-induced expression and secretion of IL-6, IL-8, and CXCL2. HMGB1, an important member of the HMG family, plays a critical role in a variety of liver diseases, such as fibrosis and tumorigenesis [Citation21,Citation22]. Notably, HBx overexpression-induced secretion of HMGB1 was inhibited by Nec-1 treatment. These findings suggest that antagonism of RIP1 with Nec-1 attenuated HBx overexpression-induced inflammation in hepatocytes.
HBx activates a variety of signalling transduction pathways in hepatocytes [Citation23]. The kinase JNK acts as a survival regulator for various cells, including hepatocytes. The activation of JNK plays a pivotal role in HBx-induced apoptosis of hepatocarcinoma cell line HepG2 cells through promoting AP-1 transactivation [Citation24]. HBx increased the expression of AP-1 target genes through c-Jun and c-fos induction in HepG2 cells [Citation25]. Interestingly, JNK-ROS signalling plays a pivotal role in RIP1-mediated necroptosis. Blockage of the JNK-ROS signalling has been reported to be a possible therapeutic strategy for hepatic fibrosis [Citation26]. In this study, we found that Nec-1 treatment inhibited HBx-induced JNK/AP-1 signalling. NF-κB activation is essential for inflammation and inflammation-associated cancerous changes [Citation27]. NF-κB is a well-unknown transcriptional modulator of genes involved in immune response, inflammatory reactions, and cell proliferation. NF-κB plays a central role in mediating HBx-induced pro-inflammatory cytokine expression in hepatocytes [Citation28]. Both AP-1 and NF-κB participate in hepatic inflammation, fibrosis, and cancer [Citation27]. The inhibitory effects of Nec-1 against HBx-induced AP-1 and NF-κB activation suggest that RIP1 might be considered as a possible therapeutic target for the treatment of liver diseases.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Wu CC, Chen YS, Cao L, et al. Hepatitis B virus infection: defective surface antigen expression and pathogenesis. World J Gastroenterol. 2018;24:3488–3499.
- Twu JS, Robinson WS. Hepatitis B virus X gene can transactivate heterologous viral sequences. Proc Natl Acad Sci USA. 1989;86:2046–2050.
- Rawat S, Clippinger AJ, Bouchard MJ. Modulation of apoptotic signaling by the hepatitis B virus X protein. Viruses. 2012;4:2945–2972.
- Zhong L, Shu W, Dai W, et al. Reactive oxygen species-mediated c-Jun NH2-terminal kinase activation contributes to hepatitis B virus X protein-induced autophagy via regulation of the beclin-1/Bcl-2 interaction. J Virol. 2017;91:pii:e00001–17.
- Kim HJ, Kim SY, Kim J, et al. Hepatitis B virus X protein induces apoptosis by enhancing translocation of Bax to mitochondria. IUBMB Life. 2008;60:473–480.
- Luo MX, Wong SH, Chan MT, et al. Autophagy mediates HBx-induced nuclear factor-κB activation and release of IL-6, IL-8, and CXCL2 in hepatocytes. J Cell Physiol. 2015;230:2382–2389.
- Hafner A, Brandenburg B, Hildt E. Reconstitution of gene expression from a regulatory-protein-deficient hepatitis B virus genome by cell-permeable HBx protein. EMBO Rep. 2003;4:767–773.
- Vandenabeele PL, Galluzzi T, Berghe V, et al. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714.
- Humphries F, Yang S, Wang B, et al. RIP kinases: key decision makers in cell death and innate immunity. Cell Death Differ. 2015;22:225–236.
- Devin A, Lin Y, Liu ZG. The role of the death-domain kinase RIP in tumour-necrosis-factor-induced activation of mitogen-activated protein kinases. EMBO Rep. 2003;4:623–627.
- Kang TB, Yang SH, Toth B, et al. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. 2013;38:27–40.
- Galluzzi L, Kepp O, Chan FK, et al. Necroptosis: mechanisms and relevance to disease. Annu Rev Pathol. 2017;12:103–130.
- Li Y, Yang X, Ma C, et al. Necroptosis contributes to the NMDA-induced excitotoxicity in rat's cultured cortical neurons. Neurosci Lett. 2008;447:120–123.
- Yip WK, Cheng AS, Zhu R, et al. Carboxyl-terminal truncated HBx regulates a distinct microRNA transcription program in hepatocellular carcinoma development. PLoS One. 2011;6:e22888.
- Srisuttee R, Koh SS, Park EH, et al. Up-regulation of Foxo4 mediated by hepatitis B virus X protein confers resistance to oxidative stress-induced cell death. Int J Mol Med. 2011;28:255–260.
- Yang F. Post-translational modification control of HBV biological processes. Front Microbiol. 2018;9:2661.
- Shirakata Y, Koike K. Hepatitis B virus X protein induces cell death by causing loss of mitochondrial membrane potential. J Biol Chem. 2003;278:22071–22078.
- Lee YI, Hwang JM, Im JH, et al. Human hepatitis B virus-X protein alters mitochondrial function and physiology in human liver cells. J Biol Chem. 2004;279:15460–15471.
- Park S, Shin H, Cho Y. Shikonin induces programmed necrosis-like cell death through the formation of receptor interacting protein 1 and 3 complex. Food Chem Toxicol. 2013;55:36–41.
- Pan X, Cao H, Lu J, et al. Interleukin-32 expression induced by hepatitis B virus protein X is mediated through activation of NF-κB. Mol Immunol. 2011;48:1573–1577.
- Fu S, Wang J, Hu X, et al. Crosstalk between hepatitis B virus X and high-mobility group box 1 facilitates autophagy in hepatocytes. Mol Oncol. 2018;12:322–338.
- Chen R, Hou W, Zhang Q, et al. Emerging role of high-mobility group box 1 (HMGB1) in liver diseases. Mol Med. 2013;19:357–366.
- Feitelson MA, Bonamassa B, Arzumanyan A. The roles of hepatitis B virus-encoded X protein in virus replication and the pathogenesis of chronic liver disease. Expert Opin Ther Targets. 2014;18:293–306.
- Tang RX, Kong FY, Fan BF, et al. HBx activates FasL and mediates HepG2 cell apoptosis through MLK3-MKK7-JNKs signal module. World J Gastroenterol. 2012;18:1485–1495.
- Cho HK, Kim SY, Kyaw YY, et al. HBx induces the proliferation of hepatocellular carcinoma cells via AP1 over-expressed as a result of ER stress. Biochem J. 2015;466:115–121.
- Jia Y, Wang F, Guo Q, et al. Curcumol induces RIPK1/RIPK3 complex-dependent necroptosis via JNK1/2-ROS signaling in hepatic stellate cells. Redox Biol. 2018;19:375–387.
- Elsharkawy AM, Mann DA. Nuclear factor-kappaB and the hepatic inflammation–fibrosis–cancer axis. Hepatology. 2007;46:590–597.
- Mozer-Lisewska I, Kaczmarek M, Zeromski J. The role of NF-kappaB transcription factor in chronic viral hepatitis C and B. Postepy Biochem. 2006;52:56–61.