
Abstract
Cases of more than three primary cancers are very rare. This study analyzed the genetic susceptibility of gene polymorphisms in three patients with multiple primary malignant neoplasms and examined the possible pathogenesis. The clinical data and whole genome sequence of three patients (1 with 5 primary cancers, 1 with 4 primary cancers, and 1 with 3 primary cancers) were aligned with a series of databases. We found the three patients contained a total of seven types of malignant tumours (endometrial cancer, ovarian cancer, breast cancer, colon cancer, ureter cancer, bladder cancer and kidney cancer). It was found that the varied genes in Patient 1 (5 primary cancers) were BRIP1, FANCG, NBN, AXIN2, SRD5A2, and CEBPA. Patient 2 (4 primary cancers) had variations in the following genes: BMPR1A, FANCD2, MLH3, BRCA2, and FANCM. Patient 3 (3 primary cancers) had variations in the following genes: MEN1, ATM, MSH3, BRCA1, FANCL, CEBPA, and FANCA. String software was used to analyze the KEGG pathway of the variations in these three samples, which revealed that the genes are involved in the Fanconi anaemia pathway. Defects in DNA damage repair may be one of the causes of multiple primary cancers.
Introduction
In 1899, Billroth proposed the definition of multiple primary malignant neoplasms (MPMNs) and in 1932, Warren and Gates revised the diagnostic criteria. In 1979, Liu added new additions to the diagnostic criteria for MPMNs [Citation1]. Most scholars agree that a second primary cancer occurring within 6 months of the first primary cancer is defined as a synchronous MPMN (SMPMNs), while secondary primary cancers occurring at 6 months or later are defined as metachronous MPMN(MMPMNs). This unique tumour population for which the number of cases is much lower than that for single primary malignancies and multiple primary cancers with more than three cancers is clinically rare. Therefore, basic studies of this type of disease are limited, and some scholars hypothesize that the pathogenesis of some multiple primary cancers is associated with familial inheritance. Patients with multiple primary cancers continuously develop tumours after each tumour type enters clinical remission, during which time other malignancies develop. Why these patients continuously develop cancers but can survive for a long period remains unclear. Most studies focused on examining the polymorphisms of varied genes in single primary malignant tumours and their effects on drug metabolism [Citation2]. The pathogenesis of MPMNs, their sensitivity to treatment, and prognosis have not been widely examined. This study evaluated three patients with multiple primary cancers who mainly developed gynaecological cancers (1 with 5 primary cancers, 1 with 4 primary cancers, and 1 with 3 primary cancers) by whole-genome sequencing of the peripheral blood to analyze the polymorphisms in genes of multiple primary cancers.
Material and methods
Clinical case reports
Patient 1 is currently aged 73 years and has five tumours. In August 1994 (50 years old), she was diagnosed with poorly differentiated endometrial cancer and underwent hysterectomy. After surgery, she underwent pelvic radiotherapy and systemic chemotherapy (chemotherapy regimen not specified). In April 2005 (60 years old), the patient had Grade III transitional cell carcinoma of the right ureter and underwent surgical resection. In January 2006 (61 years old), the patient developed Grade I transitional cell carcinoma of the bladder and underwent electrical resection of the bladder. After surgery, her bladder was infused with mitomycin for chemotherapy eight times. In April 2008 (64 years old), she developed Grade II sigmoid ulcerative colon adenocarcinoma, which invaded the intestinal wall to the external fibrous tissue, without the involvement of seven lymph nodes. The patient underwent surgical treatment, followed by postoperative chemotherapy with the FOLFOX4 regimen for eight cycles. In October 2013 (69 years old), the patient developed right invasive ductal carcinoma breast cancer (Luminal B) and underwent modified radical mastectomy of the right breast. After surgery, she underwent chemotherapy with the DE regimen (Docetaxel 100 mg day 1 + Epirubicin 80 mg day 1) for six cycles. Postoperative pathological comparison of the five tumours was carried out to exclude metastasis and revealed primary tumours. After chemotherapy for breast adenocarcinoma was ended, the patient was followed up once every six months and was still alive when follow-up ended. The patient's father and first older brother had gastric cancer, her second older brother was diagnosed with three primary cancer, respectively, colon cancer, squamous cell carcinoma of the skin and prostate cancer, her niece had leukaemia. The father and two older brothers have already died ().
Figure 1. Analytical process for genetic susceptibility of three patients.
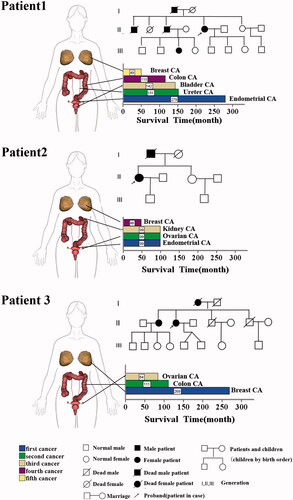
Patient 2 is a 55-year-old patient who developed four types of tumours. In August 2009 (47 years old), she was diagnosed with endometrial cancer, ovarian serous cystadenocarcinoma, and clear cell renal cell carcinoma of the left kidney. She underwent treatment with radical hysterectomy, bilateral salpingo-oophorectomy, omentectomy, pelvic lymphadenectomy, appendectomy, and left nephrectomy. After surgery, pathological comparison was carried out by immunohistochemistry and the patient was diagnosed with three synchronous primary cancers. After surgery, the patient underwent paclitaxel + oxaliplatin treatment for a total of 10 cycles. In November 2013 (51 years), the patient was diagnosed with left invasive ductal carcinoma breast cancer (Luminal B) and underwent modified radical mastectomy. After surgery, pathological comparison was carried out with her previous surgery to exclude metastasis and primary breast adenocarcinoma was considered. After surgery, the patient underwent the EC-TH regimen for eight cycles (cyclophosphamide 1.1 g day1 + epirubicin 110 mg day 1 q3w ×4 cycles, sequential trastuzumab 440 mg day 1 + docetaxel 130 mg d2 q3w × 4 cycles, followed by herceptin as maintenance therapy for up to 1 year). After chemotherapy, the patient went for follow-up once every six months and was still alive at the end of follow-up. The patient's father died of lung cancer at the age of 87 ().
Patient 3 is a 76-year-old patient who developed three types of tumours. In June 1995 (54 years old), the patient was first diagnosed with left invasive ductal breast cancer (Luminal, A/B cannot be determined as c-erbB2 and ki67 were not tested). The patient underwent modified radical mastectomy of the left breast. After surgery, the patient underwent adjuvant chemotherapy with the CEF regimen for 10 cycles, radiotherapy to the chest wall, and tamoxifen endocrine therapy. In August 2008 (68 years old), she developed Grade I cecal adenocarcinoma which invaded to the mucosal and muscle layer. The patient underwent radical surgery for colon cancer and pathology tests were carried out after surgery to exclude breast adenocarcinoma metastasis. After surgery, the patient underwent chemotherapy for six cycles with the mFOLFOX regimen. In November 2010 (71 years), the patient developed grade III right ovarian serous cystadenocarcinoma and underwent radical hysterectomy and bilateral salpingo-oophorectomy. After surgery, pathological comparison was carried out with the previous two pathological results to confirm that the patient had primary ovarian cancer. After surgery, the patient underwent six cycles of systemic chemotherapy with the DC regimen (docetaxel 120 mg day1 + Carboplatin 400 mg day1 q3w ×6 cycles). In 2014 (74 years old), a review showed continuous CA-125 elevation accompanied by peritoneal effusion. The patient successively underwent peritoneal perfusion with cisplatin and systemic chemotherapy with the DC regimen a total of 11 cycles in two years. The patient was alive when follow-up ended. The patient's mother and older sister all had breast cancer and her mother has already died ().
Patient data and samples
Patient data and blood specimens were from the cancer centre of the First Affiliated Hospital of Xi’an Jiaotong University (Xi’an, China). The diagnostic principles of MPMN are based on the following standards [Citation1]: (1) each tumour is malignant, (2) each tumour has its own pathological features, (3) tumours occur in different parts or organs, and are not continuous with each other, and (4) each tumour has its own metastatic pathway and the diagnosis of metastatic or recurrent tumours can be excluded. All three patients had a confirmed diagnosis of MPMN based on standards, and clinicopathological examinations were confirmed by two experienced pathologists of the First Affiliated Hospital of Xi'an Jiaotong University (Xi’an, China). Two millilitres of blood was drawn from each patient and stored with EDTA as an anti-coagulant. After sample collection, samples were rapidly stored in a −80 °C freezer until use. Collection of patient data, experimental sampling, and whole genome sequencing were carried out with the informed consent of the patients. All methods were carried out in accordance with relevant guidelines and regulations. All experimental protocols were approved by the First Affiliated Hospital of Xi'an Jiaotong University and Ethics Committee (Xi’an, China).
Methods
Clinical data
The clinical data of the three patients with MPMNs were retrospectively analyzed. The clinical data included the interval between each type of tumour, treatment status, and survival status during follow-up of the patients. The survival time is the period from confirmed diagnosis of the tumour to death or end of follow-up. The end of follow-up of all three patients in this study was November 6, 2017 and no patients were lost to follow-up. At the end of follow-up, all three patients were still alive. SPSS 20.0 software (SPSS, Inc., Chicago, IL) was used for statistical analysis of clinical database.
Genomic DNA extraction and testing
After total DNA from the patients was used for preliminary examination, the samples were reviewed by Novogene Co., Ltd. (Beijing, China). Samples which passed review were used to construct DNA libraries and sequenced using an Illumina HiSeq X Ten (San Diego, CA).
The QIAamp blood midi kit (Qiagen, Hilden, Germany) was used to extract genomic DNA from the blood samples. DNA sample examination was carried out using three methods: (1) agarose gel electrophoresis to analyze the level of DNA degradation and detect RNA contamination; (2) an Implen Nanodrop (Munich, Germany) was used to measure DNA purity (OD 260/280 ratio); (3) the Invitrogen Qubit assay (Carlsbad, CA) was used to precisely quantify DNA concentration. DNA samples with an OD ratio of 1.8–2.0 and amount ≥1.5 μg were used for library construction.
Mutation test based on four aspects
Whole-genome sequencing was conducted on the Illumina HiSeq X Ten platform.Mutation test based on four aspects: 1. Single nucleotide polymorphism (SNP)/single nucleotide variants (SNV). 2. Insertion and deletion (InDel). 3. Copy number variation (CNV). 4. Structural variation (SV).
SAMtools [Citation3] was used to carry out SNP(SNV)and InDel analysis on the sequencing results, and ANNOVAR software [Citation4] was used to annotate SNP and InDel loci.Control-FREEC [Citation5] was used to carry out CNV analysis, BreakDancer [Citation6] was used to carry out SV analysis.
Candidate genes selection
We implement below steps to obtain the candidate genes based on SNP and INDEL mutation across three patients.1. Filtering out dbSNP_nonflagged (including minor allele frequency (MAF) ≥1% and clinically irrelevant mutation loci). This step retained mutation loci that were not in dbSNP_nonflagged. 2. Filtering mutation loci from the 1000 Genomes Project database [Citation7] (frequency >0.005 in the human population) to obtain rare mutations that can cause disease. This step retained mutation loci in the 1000 G with a frequency <0.005. 3. Screening of mutations in exons or splice sites (10 base pairs upstream and downstream). 4. Exclusion of synonymous mutations (do not change encoded amino acids) to obtain mutations affecting gene expression products. 5. Obtaining non-synonymous mutation loci affecting protein function based on SIFT [Citation8] prediction. A mutation locus with an SIFT value <0.05 was considered to have large effects on gene function. 6. According to the target of gene polymorphisms conferring genetic susceptibility, databases were selected for alignment and screening to obtain mutated genes and loci.
Crucial genes identification and functional analysis
In this study, we extensively used the sequencing data pertaining to the variant gene selection, particularly SNP, to further understand the association between variant genes and MPMNs based on functional analysis and signalling pathway analysis. We compare variant genes of each patient with the genetic susceptibility of genes databases including Cancer Gene Census (CGC) database [Citation9] and two susceptibility gene databases [Citation10, Citation11]. Then, to better illustrate the potential functional mechanism of the varied genes(cellular process, metabolic process, single-organism process and catalytic activity [Citation12]), the Kyoto encyclopedia of genes and genomes (KEGG) pathway and gene ontology (GO) functional enrichment analysis were carried out using the database for annotation, visualization and integrated discovery the DAVID Bioinformatics Tool [Citation13], which consists of an integrated biological knowledge base and analytic tools aimed at systematically extracting biological meaning from large gene/protein lists. To do this, firstly, the genes involved in the functional analysis. Subsequently, construction of the patient-function network using Cytoscape v.3.8.5 software [Citation14]. Finally, STRING 10.5 software(STRING, Inc., USA) [Citation15] was used to analyze the varied genes and loci and potential signalling pathways involved.
Results
Design and analysis process of the target gene
Whole genome sequencing of three multiple primary cancers was obtained. The design and analysis process for obtaining of target genes was shown in .
Statistical analysis of clinical patient data
All three patients were females with a total of seven types of malignant tumours (endometrial cancer, ovarian cancer, breast cancer, colon cancer, ureter cancer, bladder cancer, and kidney cancer), for a total of 12 cancers. The first primary tumour was diagnosed at ages of 47–54 years and all tumours were gynaecological. Gynaecological tumours accounted for 58.3% of lesion sites, while urinary malignancies accounted for 25% and malignancies of the digestive tract accounted for 16.7%. All three patients developed breast cancer and all had a family history of cancer. These three patients had no history of smoking and alcohol use. All of them had childbearing. The first three tumours in patient 2 were synchronous MPMNs, and the fourth primary tumour occurred 51 months after the first three tumours. The second primary tumours of patients 1 and 3 occurred 128 and 158 months, respectively, after the first primary tumour. The remaining tumours occurred >9 months after the previous tumours and were metachronous malignancies. Each tumour type in the 3 MPMN patients were treated with combination therapy with surgery as the main method. Additionally, 83.3% of tumours were treated with postoperative chemotherapy. (, ).
Figure 2. Clinical information and pedigree of three cases of MPMNs. There are three parts, the left side is the site of the disease of three multiple primary cancer patients, which are represented by the 3 D body graph, and the arrow points to the lower right, which is the histogram of the survival time of each patient. The survival time is the period from the diagnosis of the tumour to death or end of follow-up. The end of follow-up of all three patients in this study was 6 November 2017. Corresponding to the upper right is the pedigree for each of the patient.
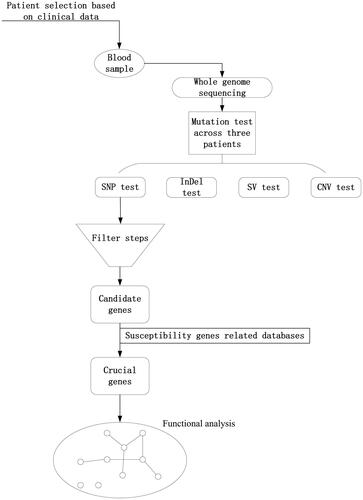
Table 1. Clinical information of three cases of MPMNs.
Table 2. The quality of sequencing data.
Quality evaluation of sequencing data
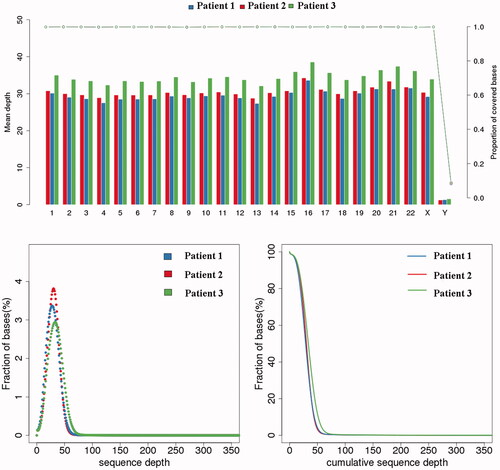
The genomic DNA was determined to be qualified for constructing DNA libraries and sequencing as shown in . Paired-end sequencing data were used for filtration and the ratio of clean reads to raw reads for subsequent bioinformatics analysis was 98.7%. The mean error rate was below 0.1% and the average ratios of Q20 and Q30 were ≥91% and ≥81%, respectively. The coverage of the sequencing data is over 99.8% and the depth is about 30X. The data of sequencing was highly reliable (, ).
Figure 3. Gel electropherogram and the quality of sample DNA for Sequencing.
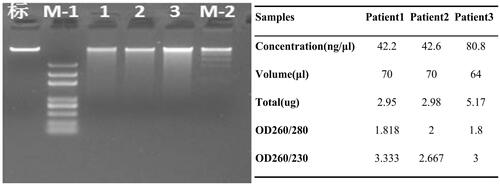
Figure 4. Quality evaluation of sequencing data. The upper graph indicates the coverage depth (left coordinate) and coverage (right coordinate) of each chromosome. The abscissa indicates the chromosome number, the left ordinate indicates the average coverage depth, and the right ordinate indicates the coverage. When calculating the depth of coverage for each chromosome, the formula is: Sequencing data volume/The total length of per chromosome. When calculating coverage, the formula is: the total length covered/the total length of each chromosome. The two images below show the depth of sequencing. The left panel shows the base ratios for different sequencing depths, the abscissa indicates the depth of sequencing, and the ordinate indicates the proportion of bases with a depth of x in all bases; the image is generally distributed around the average depth. The graph on the right shows the cumulative base ratio at different depths. The abscissa indicates the depth of sequencing. The ordinate indicates the proportion of bases with a depth greater than x in all bases. For example, the ratio of bases with a depth of 0X corresponds to 100%. Indicates that there are 100% bases with a sequencing depth greater than 0X.The results show that the coverage of the sequencing data is over 99.8% and the depth is about 30×.
Mutation test
We got the data of mutation test for each sample. Number of all SNV and InDel were about 3093792 (3090015–3097548) and 400902 (395488–406744); number of new SNV and new InDel not annotated with dbSNP were about 40706 (40632–40878) and 282580(278021–288337); number of loss in CNV were about 94 (74–120), and number of gain were about 118 (89–140); The number of deletions, insertions, inversions, and translocations in SV were approximately 1242 (1125–1356), 31 (13–46), 91 (86–106), and 381 (321–487), respectively ().
Table 3. The mutation test of sequencing data.
Candidate genes of three patients
A total of 4,918,536 SNP sites and 637,079 INDEL sites were obtained by comprehensive analysis. 119,145 SNP sites and 454,856 INDEL sites were obtained by dbSNP_nonflagged filtering in step one. 117,564 SNP mutations (rare) that can cause disease and 453,443 INDE sites were obtained by Filtering mutation loci from the 1000 Genomes Project database in step two. 1143 SNP sites that can affect gene expression products and 994 INDEL sites were obtained by screening of mutations in exons or splice sites in step three.773 SNP mutations affecting gene expression products were obtained by excluding of synonymous mutations in step four. Finally, 376 SNPs are obtained by step five based on SIFT prediction.
Crucial genes
The variation was aligned with the database which was associated with cancer inheritance, retaining the results shared by the database. In patient 1, we detected seven varied genes: BRIP1, FANCG, NBN, AXIN2, SRD5A2, and CEBPA. Patient 2 had five varied genes: BMPR1A, FANCD2, MLH3, BRCA2, and FANCM. Patient 3 had seven variations: MEN1, ATM, MSH3, BRCA1, FANCL, CEBPA, and FANCA. The results are shown in .
Table 4. List of polymorphism selected from three samples.
Functional analysis
To further characterize the genes, KEGG analyses were used to investigate their potential involvement in biological processes associated with MPMN development. We performed functional analysis using the varied genes of three patients in each patient module. The three patients related to functions network (, ) using Cytoscape v.3.8.5. Results showed that the patient 1 related to more function than others, We also found that phospholipid transporter activity and ATPase activity (coupled to transmembrane movement of substances and coupled to movement of substances) played an important role in three MPMNs.
Figure 5. The patient-function network analysis. The circle represents the function of the variant gene. The more overlapping the function, the larger the circle.
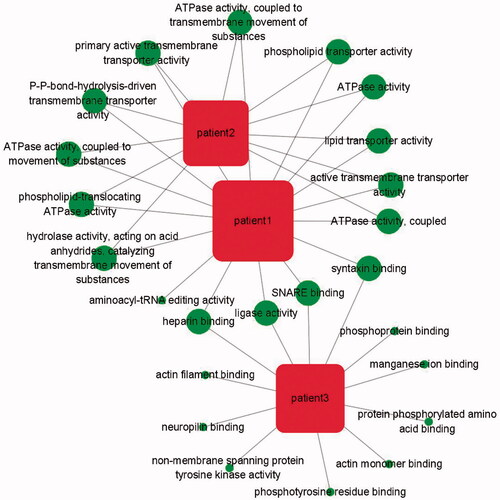
Table 5. The functional analysis of crucial genes.
String 10.5 software was used to analyze the signalling pathways of the genes containing the variations (). Genes containing the BPIP1 and FANCG variations in patient 1, FANCD2, BRCA2, and FANCM variations in patient 2, and FANCL and FANCA variations in patient 3 are involved the Fanconi anaemia pathway. The MLH3 and BRCA2 genes form a network through BRCA2, while the ATM, MSH, and BRCA1 genes and Fanconi anaemia pathway form a network through BRCA1. These genes are involved in the DNA mismatch repair system. MEN1, ATM and CEBPA variations in patient 3 are involved the pathway of transcriptional misregulation in cancer. Overall, all variations were detected in genes involved in the Fanconi anaemia pathway to form a complex network system (.
Figure 6. KEGG pathway of three samples. The top three graphs show the KEGG pathway for each patient. The graph at the bottom left shows network of all the post-screening variations involved. Overall, all variations were detected in genes involved in the FA/BRCA pathway to form a complex network system.
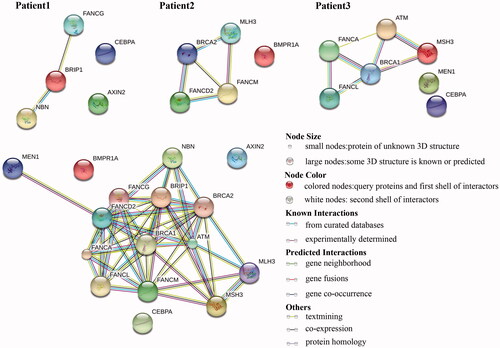
Table 6. KEGG pathway of crucial genes in three samples.
Discussion
The reported incidence of MPMN is 0.73–11.7% [Citation16]. Most studies statistically analyzed clinical cases. Patients with more than three primary malignancies are clinically rare and patients with a genetic background of malignancies may be MPMN-susceptible. All three patients evaluated in this study had a family history of cancer. Patient 3’s mother and older sister are breast cancer patients, and thus, this patient has a higher risk of hereditary breast and ovarian cancers. Patient 1 has a family history of different types of tumours and the patient herself had five types of tumours. Her second older brother developed three types of tumours. Other members of family had different single primary tumours. The pathogenic factors are complex and it is difficult to use a single tumour susceptibility gene to explain all types of tumours in the patient. Family members of Patient 2 are fewer and the patient herself had four types of tumours, only dating back to her father having had lung cancer. It is also difficult to infer the relevance of genetic pathogenesis. Second-generation whole genome detection technology enables evaluation of the pathogenic causes of these complex diseases.
Based on the screening results of this study, the onset of disease in patient 3 was closely related to nonsense mutations in BRCA1 (rs397508983) (exon9:c.C2431T:p.Q811X, exon10:c.C2572T:p.Q858X). For patient 2, the loci of interest are FANCD2 (exonicc.G983A p.R328Q), MLH3 (exonic c.C2825T p.T942I), and CEBPA (exonic c.546_548delp.182_183del). For patient 1, the loci of interest are FANCG (exonic c.A821Gp.K274R), NBN (exonic c.C1809A p.F603L), AXIN2 (exonic c.T1219C p.S407P), SRD5A2 (exonicc.C699Gp.F233L), and CEBPA (exonicc.568_569insCGCACC p.S190delinsSHP). Based on these screening results, it is difficult to determine whether tumours in patients 1 and 2 were caused by genetic variations. The aetiology of only one patient can be explained based on genetic susceptibility. Through functional analysis, we found that there are some close associations between the function genes of the three patients, but the pathogenesis of MPMNs cannot be speculated. However, signalling pathway analysis revealed that gene variations in all samples were associated with the unique Fanconi anaemia pathway, suggesting that these patients have similar causes of disease onset.
Fanconi anaemia (FA) is an autosomal recessive inherited disease and involves hematopoietic stem cells. Patients with this disease are at a high risk of progression into acute leukemia and solid malignancies and thus this disease has been widely evaluated. It has been reported that inherited homozygous (bi-allelic) mutations of Fanconi anaemia pathway can cause Fanconi anaemia (FA) and increase susceptibility to both haematologic and non-haematologic malignancies, while Heterozygous FA gene (mono-allelic) mutations somatically do not cause FA phenotype but significantly increase cancer susceptibility in a sporadic manner [Citation17]. Currently, it is known that the FA gene includes at least 16 subtypes [Citation18] and the protein encoded by these genes is intimately associated with the protein encoded by the BRCA genes to form a complex regulatory pathway. This pathway participates in cell cycle regulation, regulation of DNA damage repair, cell growth and differentiation, malignant transformation of cells, and the formation and development of cancer cells [Citation19]. BRCA genes are well-known tumour suppressor genes and mutations in this family of genes can result in high susceptibility to breast cancer and ovarian cancer. BRCA1 negatively regulates the cell cycle [Citation20], and it is known that BRCA and FANCA function together [Citation21]. Previous studies showed that FANCJ and BACH1 (BRCA1-associated C-terminal helicase) [Citation22], FANCD1 and BRCA2, FANCN and PALB2, FANCJ and BRIP1 are the same proteins [Citation23], demonstrating that FA genes and the BRCA gene family are strongly related. These multiple FA proteins and BRCA proteins form a complex functional network-FA/BRCA pathway that participates in DNA damage repair in the body.
The three patients had a total of seven types of cancer, which not only included breast cancer, ovarian cancer, and endometrial cancer, but also colon cancer and urinary system malignancies. Previous studies reported that three gene mutations in the FA/BRCA pathway result in heterozygous mutations that increase the susceptibility to breast cancer [Citation18], and dysregulation of this pathway can cause ovarian cancer [Citation24]. However, while there are no relevant studies of the FA/BRCA pathway and endometrial cancer, some reports suggested that breast cancer patients with BRCA2 mutations develop secondary endometrial cancer, suggesting that both cancers are related to the FA/BRCA pathway [Citation25]. Some articles also reported that primary early-onset colorectal cancers is associated with FANCD1/BRCA2 biallelic mutations [Citation26]. Coincidentally, the patient reported in the article had MPMN, while patients with both patient 1 and patient 2 develop colon cancer, suggesting that this pathway is related to the occurrence of colon cancer. Another previous study suggested that FA/BRCA pathway dysregulation is conducive to the development of bladder cancer [Citation27]. The FA/BRCA pathway not only plays an important role in gynaecological cancers, but FA gene mutations also occur in head and neck squamous cell carcinoma, oesophageal cancer, liver cancer, skin cancer, brain cancer, kidney cancer, and other solid malignancies [Citation18]. A team from University of Barcelona detected an enrichment for variants in FA DNA damage repair pathway genes in the familial colorectal cancer (CRC) cohort as six families carried heterozygous, rare, potentially pathogenic variants located in BRCA2/FANCD1, BRIP1/FANCJ, FANCC, FANCE and REV3L/POLZ. Their results suggest that the FA DNA damage repair pathway may play an important role in the inherited predisposition to CRC [Citation28]. These studies mostly analysed single primary malignancies, while no studies have focused on MPMNs and the FA/BRCA pathway. Although we cannot directly determine the aetiology of these three patients based on our screening results, disease onset appears to be associated with the FA/BRCA pathway and may involve DNA damage repair.
DNA damage can be caused by natural hydrolysis and different physical (UV, X-rays) or multiple chemical reagents, including reactive oxygen species produced from cellular metabolism. DNA damage alters cellular function, triggers cellular senescence or cell death, or causes cancer or ageing [Citation29]. Defects in DNA repair result in abnormal expression of some genes, affecting DNA transcription and replication. This may result in abnormal cell proliferation, which is typically referred to as tumorigenesis. In the FA/BRCA pathway, the FA protein complex interacts with and participates in homologous recombination. Defects in the expression of any gene product can result in defects in DNA damage repair. This will result in increased levels of DNA mismatch mutations and malignant transformation of normal cells. The FA/BRCA pathway is associated with DNA damage repair, and the complex network relationships are intimately associated with the cell cycle and other oncogenic pathways which affect disease outcome. The gene variations in this study are all involved in the FA/BRCA pathway, and thus, defects in this DNA repair mechanism may be among the causes of MPMNs.
The three patients have many common features. All had family history of cancer. All had MPMNs mainly with gynaecological cancers and all developed breast cancer. The patients received combined treatment with surgery as the main therapy for each disease and most cancers were treated with adjuvant chemotherapy. The patients achieved ideal therapeutic results for each episode of cancer and had a relatively long survival period. Studies of breast cancer have shown that patients with the molecular subtype of luminal B can benefit from postoperative chemotherapy. Two patients in this study (patients 1 and 2) had luminal B breast cancer, while the molecular subtype of patient 3 was unclear. Whether patients with this molecular subtype are associated with the FA/BRCA pathway requires further examination. The FA/BRCA pathway can regulate DNA damage repair in the body, and defects in expression of genes in this pathway increase genome instability in FA patients and promote cancer occurrence [Citation30]. In contrast, various subtypes in the FA family are all sensitive to DNA cross-linking agents such as mitomycin [Citation31], suggesting that all FA proteins elicit their effects through a common pathway. This change enables patients to benefit from chemotherapy and may be one of the reasons for treatment sensitivity in these patients.
The longer survival time of patients may be related to the use of combined treatment with surgery as the main treatment. These patients were all treated with appropriate chemotherapy and showed good treatment outcomes because of treatment sensitivity. At the gene level, we hypothesized that gene variations associated with the FA/BRCA family result in dysregulation of the DNA mismatch repair system and cause patients to easily develop tumours. However, patients with gene variations in the FA family show a heightened sensitivity to chemotherapeutic agents because of their genetic status, resulting in better therapeutic efficacy and increasing their survival time. Longer survival enables tumour susceptibility genes to reactivate the FA/BRCA pathway and other related signalling pathways to induce tumours, resulting in the formation of secondary malignancies.
The sample size of this study was small and the use of databases for alignment with whole-genome sequencing results may not have reflected the whole spectrum of genetic susceptibility polymorphisms. The analysis, deduction, and conclusions from the data may only represent characteristics of MPMN patients showing similar features. Because the incidence of MPMNs is low and most studies of this disease have focused on clinical analysis, few in-depth molecular biology studies of this disease have been conducted. More meaningful data can be obtained by genetically testing a greater number of patients to evaluate the pathogenesis of MPMN, which will help in the treatment of these patients.
Cases of more than three primary cancers are very rare. This study analyzed the genetic susceptibility of gene polymorphisms in patients with multiple primary malignant neoplasms and examined the possible pathogenesis. It was found that the varied genes in patients which revealed that the genes are involved in the Fanconi anaemia pathway. Defects in DNA damage repair involved in the Fanconi anaemia pathway may be one of the causes of multiple primary cancers.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Acknowledgements
The authors thank Zheling Chen and Yisong Xu for excellent technical assistance.
Disclosure statement
The authors report no conflict of interest.
Data availability
All data generated or analysed during this study are included in this published article
Additional information
Funding
References
- Xu LL, Gu KS. Clinical retrospective analysis of cases with multiple primary malignant neoplasms. Genet Mol Res. 2014;13:9271–9284.
- Xu Y, Zhang C, Liang H, et al. Dishevelled 1, a pivotal positive regulator of the Wnt signalling pathway, mediates 5-fluorouracil resistance in HepG2 cells. Artif Cells Nanomed Biotechnol. 2018;46:1–9.
- Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079.
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
- Boeva V, Popova T, Bleakley K, et al. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics. 2012;28:423–425.
- Chen K, Wallis JW, McLellan MD, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–681.
- Buchanan CC, Torstenson ES, Bush WS, et al. A comparison of cataloged variation between International HapMap Consortium and 1000 Genomes Project data. J Am Med Inform Assoc. 2012;19:289–294.
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812.
- Sondka Z, Bamford S, Cole CG, et al. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat Rev Cancer. 2018;18:696–705.
- Rahman N. Realizing the promise of cancer predisposition genes. Nature. 2014;505:302–308.
- Gast AC, Metzger J, Tipold A, et al. Genome-wide association study for hereditary ataxia in the Parson Russell Terrier and DNA-testing for ataxia-associated mutations in the Parson and Jack Russell Terrier. BMC Veterinary Res. 2016;12:225.
- Wen S, Wang X, Wang Y, et al. Nucleoside diphosphate kinase 2 confers acquired 5-fluorouracil resistance in colorectal cancer cells. Artif Cells, Nanomed Biotechnol. 2018;23:1–10.
- Jiao X, Sherman BT, Huang da W, et al. DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics. 2012;28:1805.
- Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504.
- Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;4;45:D362–D368.
- Demandante CG, Troyer DA, Miles TP. Multiple primary malignant neoplasms: case report and a comprehensive review of the literature. Am J Clin Oncol. 2003;26:79–83.
- Stecklein SR, Jensen RA. Identifying and exploiting defects in the Fanconi anemia/BRCA pathway in oncology. Transl Res. 2012;160:178–197.
- Haitjema A, Brandt BW, Ameziane N, et al. A protein prioritization approach tailored for the FA/BRCA pathway. PLoS One. 2013;8:e62017.
- Chen P, Li J, Jiang HG, et al. Curcumin reverses cisplatin resistance in cisplatin-resistant lung caner cells by inhibiting FA/BRCA pathway. Tumor Biol. 2015;36:3591–3599.
- Flower KJ, Shenker NS, El-Bahrawy M, et al. DNA methylation profiling to assess pathogenicity of BRCA1 unclassified variants in breast cancer. Epigenetics. 2015;10:1121–1132.
- Haitjema A, Mol BM, Kooi IE, et al. Coregulation of FANCA and BRCA1 in human cells. SpringerPlus. 2014;3:381.
- Litman R, Peng M, Jin Z, et al. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 2005;8:255–265.
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8:735–748.
- D'Andrea AD. The Fanconi Anemia/BRCA signaling pathway: disruption in cisplatin-sensitive ovarian cancers. Cell Cycle. 2003;2:290–292.
- Oh SE, Kim SH, Kim MS, et al. Endometrial cancer occurence five years after breast cancer in BRCA2 mutation patient. Obstet Gynecol Sci. 2015;58:175.
- Degrolard-Courcet E, Sokolowska J, Padeano MM, et al. Development of primary early-onset colorectal cancers due to biallelic mutations of the FANCD1/BRCA2 gene. Eur J Hum Genet. 2014;22:979–987.
- Neveling K, Kalb R, Florl AR, et al. Disruption of the FA/BRCA pathway in bladder cancer. Cytogenet Genome Res. 2007;118:166–176.
- Esteban-Jurado C, Franch ES, Munoz J, et al. The Fanconi anemia DNA damage repair pathway in the spotlight for germline predisposition to colorectal cancer. Eur J Hum Genet. 2016;24:1501–1505.
- Hoeijmakers JH. Genome maintenance mechanisms are critical for preventing cancer as well as other aging-associated diseases. Mech Ageing Dev. 2007;128:460–462.
- Rodriguez A, Torres L, Juarez U, et al. Fanconi anemia cells with unrepaired DNA damage activate components of the checkpoint recovery process. Theoretical Biol Med Model. 2015;12:19.
- Howlett NG, Taniguchi T, Olson S, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609.