
Abstract
Plastin 3 (PLS3) overexpression may serve as a marker for predicting chemotherapeutic outcomes in drug-resistant cancer cells, but the mechanism is unclear. Herein, we show that the down-regulation of PLS3 by PLS3 gene silencing augments the sensitivity of MDA-MB-231 triple-negative breast cancer cells to paclitaxel. Interestingly, a low concentration of paclitaxel was able to induce strong apoptosis in the PLS3-silenced cells. Further study revealed that p38 MAPK signalling was responsible for the increased sensitivity to paclitaxel in these cells, as the p38 MAPK inhibitor SB203580 impaired the changes mediated by PLS3 down-regulation in response to paclitaxel. Therefore, our study identifies PLS3 as a potential target for enhancing the p38 MAPK-mediated apoptosis induced by paclitaxel. Unlike paclitaxel, Abraxane was unable to induce strong apoptosis in the PLS3-silenced cells. As PLS3 was found to be involved in the process of endocytosis in breast cancer cells, the reliance of cellular Abraxane uptake on this process may render it not as efficient as paclitaxel in PLS3-depleted tumour cells. The finding that PLS3 could be a critical regulator of paclitaxel sensitivity may have important implications for breast cancer chemotherapy.
Introduction
Paclitaxel (PTX) is an effective anti-cancer agent for the treatment of breast cancer, ovarian cancer, lung cancer, and head and neck cancer, among other cancer types [Citation1–3]. It is also useful for treating triple-negative breast cancer (TNBC) in routine clinical practice. As TNBC lacks expression of the oestrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2), there is no preferred chemotherapy for TNBC, instead of other breast cancer subtypes. The combination of docetaxel or PTX with anthracycline agents may be more beneficial for the treatment of TNBC than for ER-positive, HER2-negative cancers [Citation4,Citation5]. Abraxane (ABR), which is the albumin-bound form of PTX, has more advantages than free PTX in that it lacks the toxicity induced by the delivery solvent Cremophor EL and has a longer circulation half-life [Citation6]. ABR (also named nab-PTX) was approved for the treatment of breast cancers after the failure of combination chemotherapy for metastatic or recurrent diseases within 6 months of adjuvant chemotherapy [Citation7]. Although PTX and ABR have improved patient survival times, the drugs are not effective in all patients with TNBC. Thus, it would be helpful to understand the mechanism behind the sensitivity of TNBC cells to PTX in order to improve the efficacy of chemotherapy with these drugs.
The PTX drugs cause tubulin disruption, induce cell cycle arrest, and finally induce DNA damage and apoptosis. It is widely accepted that PTX-induced apoptosis depends on the extracellular signal-regulated kinase and p38 mitogen-activated protein kinase (MAPK) signalling pathways [Citation8–10]. For instance, the p38 MAPK signalling pathway was activated following PTX administration, whereas a p38 MAPK inhibitor interfered with the apoptotic response [Citation10]. Some evidence has also indicated that the p38 MAPK signalling pathway is related to the PTX resistance of ovarian carcinoma and non-small-cell lung cancer [Citation11,Citation12]. Moreover, p38 MAPK played a significant role in the activation of p53, which was provoked by chemotherapeutic agents [Citation13]. p38 MAPK-mediated apoptosis in PTX-resistant ovarian carcinoma cells depended on the activation of p53 [Citation11]. When p53 was activated, it initiated a transcription program that consisted of p53-responsive proteins and genes that resulted in cell cycle arrest or apoptosis. Under stimulation, p53 decreased the gene expression of the apoptosis regulator B-cell lymphoma 2 (Bcl2) and increased the gene expression of Bcl-2-associated X protein (Bax) in mice [Citation14]. As a downstream gene of p38 MAPK, BCL2 could affect PTX-mediated apoptosis as well. The down-regulation of Bcl-2 by small interfering RNA (siRNA) significantly augmented PTX-induced apoptosis through the activation of calpain and caspases [Citation15]. Notably, PTX-induced apoptosis might be affected by altering the activities of the p38 MAPK signalling pathway, but the precise molecular mechanism involved has not been elucidated.
Herein, we report that another effector molecule, plastin 3 (PLS3), is involved in the regulation of PTX-induced apoptosis. PLS3 (a T-plastin, also named T-fimbrin) functions as filamentous actin (F-actin)-bundling protein to polymerize actin fibres by inhibiting depolymerization [Citation16,Citation17]. PLS3 plays an important role in early cancer diagnosis and therapy [Citation18,Citation19]. Since the 1980s, several studies have reported that PLS3 expression was associated with cellular resistance to chemotherapeutic drugs and cancer progression. Compared with the PLS3 expression level in cisplatin-sensitive human cancer cell lines (e.g. bladder, prostate, and head and neck cancer cell lines), that in their cisplatin-resistant counterparts was increased [Citation20]. The down-regulation of PLS3 expression increased the sensitivity not only to cisplatin in bladder cancer cells but also to VP-16 in human liver cells [Citation21]. These findings indicated that PLS3 may be a novel marker of drug resistance in cancer cells and that the relationship between its expression and drug sensitivity might be due to its involvement in apoptosis. However, the exact details behind the relevance of PLS3 expression to drug therapy and cancer progression are still unknown. In this present study, we sought to identify the role that PLS3 plays in PTX-induced apoptosis in cancer cells. To this end, we conducted PLS3 gene-silencing studies using a TNBC cell line, MDA-MB-231 and compared the responses of the gene-silenced cells under PTX and ABR exposure.
Materials and methods
Cell culture
MDA-MB-231 (human breast cancer) cells were purchased from the American Type Culture Collection and maintained as previously described [Citation22]. All cultures were routinely tested for the presence of mycoplasma and for the retention of their respective morphological and growth characteristics.
siRNA interference and transfection
T-Plastin-specific siRNA (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used to knock down the PLS3 mRNA (designated as si-PLS3). The Negative Control siRNA (Ribobio Technology, Guangzhou, China) was applied as a negative control (designated as Ncontrol). Cells at 40–60% confluence in six-well culture dishes were transfected with the respective siRNAs using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Reagents and antibodies
ABR was obtained from Celgene Co. (Summit, NJ, USA), whereas PTX was procured from the Zhejiang Hisun Pharmaceutical Factory (Taizhou, Zhejiang, China). SB203580 was purchased from MedChem Express (Monmouth Junction, NJ, USA). The primary antibodies used were as follows: T-plastin, p53, and E3 ubiquitin-protein ligase Mdm-2 (MDM-2) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); Bcl-2 was from Genetex (Irvine, CA, USA); and p-p38 MAPK, t-p38 MAPK and β-actin were from Cell Signaling Technology (Danvers, MA, USA). The secondary antibodies were purchased from Bioeasy Technology (Beijing, China).
Cytotoxicity assay
The cytotoxicity of the drugs was determined by sulforhodamine B (SRB) assay, as described previously [Citation23]. After transfection with siRNA for 24 h, the cells were seeded in 96-well plates at a density of 5 × 103 and allowed to incubate for 24 h. After an additional incubation with PTX or ABR for 48 h, the cells were fixed with 10% trichloroacetic acid for 1 h at 4 °C and then stained with 0.4% (w/v) SRB in 1% acetic acid for 30 min at room temperature. Any unbound stain was removed by washing the cells twice with 1% acetic acid. The SRB-stained cells were then solubilized with 10 mM Tris buffer (pH 10.5) (Sigma-Aldrich, St. Louis, MO, USA) and the absorbance was read at a wavelength of 570 nm.
Western blot analysis
After the siRNA transfection and drug treatments, the cells were harvested and lysed in RIPA buffer (Cell Signaling Technology, Danvers, MA, USA) and the protein concentration was determined using an Enhanced BCA Protein assay kit (Beyotime, Shanghai, China). The total cell lysates were subjected to SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore, Burlington, MA, USA). The membranes were incubated overnight at 4 °C with the indicated antibodies at 1:500 dilution (antibodies from Santa Cruz Biotechnology) or 1:1000 dilution (antibodies from Genetex and Cell Signaling Technology, Danvers, MA, USA). Then, they were incubated for 1 h at room temperature with the horseradish peroxidase-conjugated secondary antibodies at 1:2000 dilution. The immunoreactive bands were developed using the enhanced chemiluminescence detection system (Millipore, Burlington, MA, USA) and detected with the ChemiDoc MP imaging system (Bio-Rad, Hercules, CA, USA).
Endocytosis and apoptosis assays
The endocytosis assay was performed using flow cytometry. Cells were transfected with the indicated siRNAs for 48 h and then incubated with 1 mg/mL fluorescein isothiocyanate (FITC)-dextran (MW 70,000; Sigma-Aldrich, USA) at 4 °C or 37 °C. At the specific time points of interest, endocytosis was stopped by washing the cells twice in ice-cold phosphate-buffered saline and the cells were then collected for subsequent flow cytometric analysis.
The cell apoptosis assay was also performed using flow cytometry. After siRNA transfection and incubation with the indicated reagents, a total of 1 × 106 cells were harvested and treated with the reagents supplied in the Annexin V-APC/propidium iodide (PI) apoptosis detection kit (Keygentec, Nanjing, China) according to the manufacturer’s protocol. The cells were collected for subsequent flow cytometric analysis and the data were analyzed using FlowJo software (version 10.0; Tree Star, Ashland, OR, USA).
Statistical analysis
Data are presented as the mean ± standard deviation (SD) from triplicate experiments. The data were analyzed with the Student t-test, where p values of less than .05 were considered significant. All statistical analyses were performed using the Statistical Package for the Social Sciences software (version 17.0; SPSS, Chicago, IL, USA).
Results
PLS3 down-regulation alone fails to impair cell proliferation but slightly inhibits apoptosis
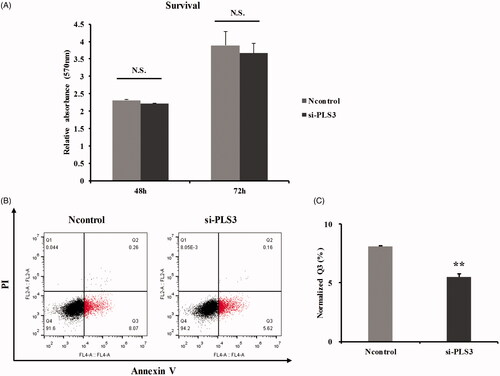
To better determine the effects of PLS3 down-regulation on the cell response to the PTX agents, we first examined the influence of PLS3 gene silencing on cell proliferation and apoptosis. PLS3-specific siRNA was, therefore, used to decrease the expression of PLS3. As shown in , PLS3 down-regulation did not result in any significant difference in the cell survival rate relative to that in the Ncontrol cells at 48 or 72 h. Then, the role of PLS3 in mediating cell death was assessed by Annexin V-APC/PI apoptosis assay. As shown in , the down-regulation of PLS3 slightly inhibited apoptosis (5.50 ± 0.27% vs. 8.09 ± 0.08% in Ncontrol cells). Thus, although PLS3 down-regulation did not alter cell proliferation, it had a slight inhibitory effect on apoptosis.
Figure 1. Down-regulation of plastin 3 (PLS3) alone fails to impair proliferation but slightly inhibits apoptosis. (A) The cell survival rate was determined by sulforhodamine B assay, and cells were transfected with Ncontrol or si-PLS3 for 24, 48 and 72 h. The relative absorbance was normalized to the value at 24 h. The histogram plots presented no significant differences in cells transfected with si-PLS3 versus Ncontrol. N.S.: No significance. (B) Representative dot plots showing apoptosis in cells transfected with Ncontrol or si-PLS3 for 48 h. Apoptotic cells were stained with Annexin V-APC/PI and then analyzed by flow cytometry. (C) Quantification of the Q3 population and histogram plots showing a significant decrease in apoptosis upon PLS3 down-regulation. The analysis was carried out using FlowJo 10.0 software. **p < .01 versus Ncontrol. All the data represent the means ± SD of three independent experiments.
PLS3 down-regulation augments the sensitivity to PTX and ABR
Since PLS3 played a role in the resistance to cisplatin in different cancer cells, we hypothesized that PLS3 expression might be associated with the cytotoxicity of other chemotherapeutics like PTX and ABR. To investigate whether PLS3 down-regulation could influence the cytotoxicities of PTX and ABR, the SRB assay was conducted on MDA-MB-231 cells transfected with Ncontrol and si-PLS3. From comparison of the half-maximal inhibitory concentration (IC50) values of PTX and ABR on the two groups of transfected cells, we found that the down-regulation of PLS3 had significantly decreased the IC50 value of PTX (2.38 ± 0.19 nM vs. 6.83 ± 0.30 nM for Ncontrol cells), but hardly had any effect on that of ABR (). When exposed to various PTX concentrations, the si-PLS3-transfected cells presented lower survival rates than those of the Ncontrol group (). Interestingly, the si-PLS3 group showed lower survival rates than those of the Ncontrol group with lower concentrations of ABR but showed similar and even higher survival rates under higher concentrations of the drug (). Thus, the down-regulation of PLS3 had varying effects on cell survival in response to PTX and ABR, where it generally augmented the cell’s sensitivity to PTX. To explore the mechanisms behind different effects of PLS3 down-regulation on cell survival in response to PTX and ABR, we used 6 nM of PTX and ABR in the present study.
Figure 2. Down-regulation of plastin 3 (PLS3) augments the cell’s sensitivity to paclitaxel (PTX) and Abraxane (ABR). The cell survival rate was determined by sulforhodamine B assay, and cells were treated with the indicated doses of PTX (A) or ABR (B) for 48 h. (C) Representative dot plots showing apoptosis in cells treated with 6 nM PTX or ABR after incubation for 6 h. Apoptotic cells were stained with Annexin V-APC/PI and then analyzed by flow cytometry. (D) Quantification of the Q3 population and histogram plots showing an increase in apoptosis upon PLS3 down-regulation. The analysis was carried out using FlowJo 10.0 software. ***p < .001, **p < .01, *p < .05. All the data represent the means ± SD of three independent experiments.
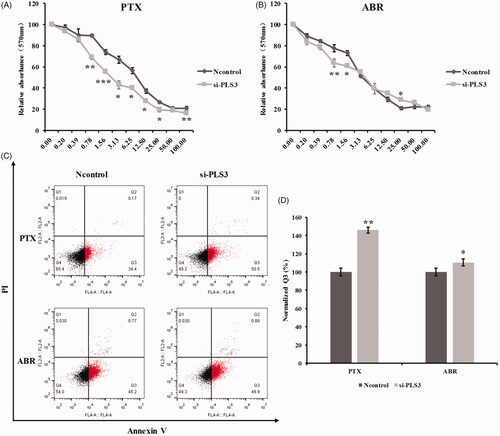
Table 1. IC50 Values in plastin 3 (PLS3)-down-regulated and control MDA-MB-231 cells treated with paclitaxel (PTX) and abraxane (ABR).
We also used the Annexin V-APC/PI apoptosis assay to assess the influence of PLS3 down-regulation on the apoptosis induced by PTX and ABR. As shown in , PLS3 down-regulation significantly augmented PTX-induced apoptosis. However, unlike its effect on cell survival, PLS3 down-regulation improved the apoptosis induced by ABR, albeit not to the extent observed with PTX treatment. Collectively, the results revealed that PLS3 down-regulation improves the cellular apoptosis induced by PTX and ABR, which may result in enhancement of the cell’s sensitivity to PTX in particular.
PLS3 down-regulation activates the p38 MAPK signalling pathway with exposure to PTX only
Next, we investigated how PLS3 down-regulation could regulate the cell’s sensitivity to the PTX drugs. We first detected the influence of PLS3 down-regulation on cell cycle. As previously reported, low concentration of PTX (6 nM) could preferentially arrest cells in G1 and G2/M by comparing the Ncontrol with the Ncontrol/PTX group (Figure S1A & B) [Citation24,Citation25]. Without drug treatment, the PLS3 down-regulation also induced G1 and G2/M arrested by comparing the si-PLS3 with the Ncontrol group, indicating that PLS3 might act as a regulator of the cell cycle. Nevertheless, the cells in the Ncontrol or si-PLS3 group displayed similar distributions of the cell cycle with low concentrations of PTX exposure, albeit the strong augment in apoptosis induced by PTX in PLS3-silenced cells. It was reported that apoptosis induced by low concentrations of paclitaxel occurred independent of mitotic arrest [Citation26]. Thus, we supposed that PLS3 down-regulation enhanced the PTX-induced apoptosis independent of the cell-cycle regulation. We then focused on the p38 MAPK signalling pathway and the downstream effector molecules MDM-2, p53 and Bcl-2, which play vital roles in PTX-induced apoptosis. As shown in , PLS3 silencing with si-PLS3 alone decreased the phosphorlyation of p38 MAPK relative to the Ncontrol level, indicating that activation of the p38 MAPK signalling pathway had been decreased. The expression of p53 was also decreased with PLS3 down-regulation. However, PTX exposure reversed these effects, where the phosphorylation of p38 MAPK was strongly promoted in the si-PLS3-transfected cells. Moreover, exposure to PTX decreased the expression of MDM-2 and Bcl-2 and increased the expression of p53 in the si-PLS3-transfected cells. Activation of p38 MAPK signalling could induce the rapid degradation of MDM-2 via ubiquitin-proteasomal degradation and enhance the stability of p53 in PTX-resistant cells [Citation12]. Here, PLS3 down-regulation might have improved the stability of p53 via the p38 MAPK-induced suppression of MDM-2 with the PTX treatment and then decreased the expression of the anti-apoptotic Bcl-2 protein. We also examined the cleavage of caspase-3, another marker of apoptosis. Similarly, PTX induced a marked increase in cleaved caspase-3 (Figure S2). Since the down-regulation of PLS3 could significantly enhance PTX-induced apoptosis, we hypothesized that this enhancement was via the p38 MAPK signalling pathway.
Figure 3. Down-regulation of plastin 3 (PLS3) activates the p38 MAPK signalling pathway with exposure to paclitaxel (PTX) but not with Abraxane (ABR). (A) The expression of PLS3, p-p38 MAPK, t-p38 MAPK, p53, MDM-2, and Bcl-2 in MDA-MB-231 cells treated with 6 nM PTX or ABR for 24 h was detected by Western blotting. MDA-MB-231 cells were transfected with Ncontrol or si-PLS3 for 48 h before treatment. β-Actin was used as a loading control. (B) Histogram plots representing the ratio of p-p38/t-p38. The expression of p-p38 and t-p38 was analyzed using ImageJ software. (C) Relative expression levels of PLS3, p53, MDM-2, and Bcl-2, normalized to the expression of β-actin. The PLS3, p53, MDM-2, and Bcl-2 expression values were analyzed using ImageJ software. *p < .05. All the data represent the means ± SD of three independent experiments.
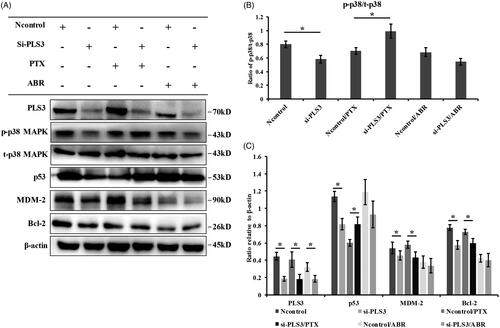
In contrast, ABR exposure impaired the effect triggered by the down-regulation of PLS3, where the si-PLS3 group revealed no significant difference compared with the Ncontrol group in the phosphorylation of p38 MAPK. The expression levels of the MDM-2, p53 and Bcl-2 proteins also did not vary significantly between the two groups of transfected cells. Combined with the results of , it would appear that the mechanism behind the enhancement of ABR-induced apoptosis by PLS3 down-regulation might have little to do with the p38 MAPK signalling pathway.
The increased cell sensitivity to PTX by PLS3 down-regulation depends on the p38 MAPK signalling pathway
We further verified whether p38 MAPK signalling could regulate the enhancement of PTX-induced apoptosis triggered by PLS3 down-regulation. To this end, we used a p38 MAPK inhibitor (SB203580) to suppress the activation of the p38 MAPK signalling pathway. We observed that the co-treatment with SB203580 and PTX impaired PTX-induced apoptotic death in cells transfected with si-PLS3, where the combination almost neutralized apoptosis compared with exposure to PTX, alone (). The result verified that PLS3 down-regulation augmented PTX-induced apoptosis via the p38 MAPK signalling pathway.
Figure 4. The increased cell sensitivity to paclitaxel (PTX) triggered by plastin 3 (PLS3) down-regulation is dependent on the p38 MAPK signalling pathway. (A) Representative dot plots showing apoptosis in cells treated with DMSO or 10 μM SB203580 or 6 nM PTX or both after incubation for 6 h. Apoptotic cells were stained with Annexin V-APC/PI and then analyzed by flow cytometry. (B) Quantification of the Q3 population and histogram plots showing a decrease in apoptosis upon exposure to SB203580 and PTX, compared with PTX alone in si-PLS3 groups. The analysis was carried out using FlowJo 10.0 software. (C) The expression of PLS3, p53, MDM-2, and Bcl-2 in MDA-MB-231 cells treated with either DMSO, 10 μM SB203580 or 6 nM PTX or both for 24 h was detected by Western blotting. The MDA-MB-231 cells were transfected with Ncontrol or si-PLS3 for 48 h before treatment. β-Actin was used as a loading control. (D) Relative expression levels of PLS3, p53, MDM-2, and Bcl-2, normalized to the expression of β-actin. The PLS3, p53, MDM-2, and Bcl-2 expression levels were analyzed using ImageJ software. ***p < .001, **p < .01, *p < .05. N.S.: No significance. All the data represent the means ± SD of three independent experiments.
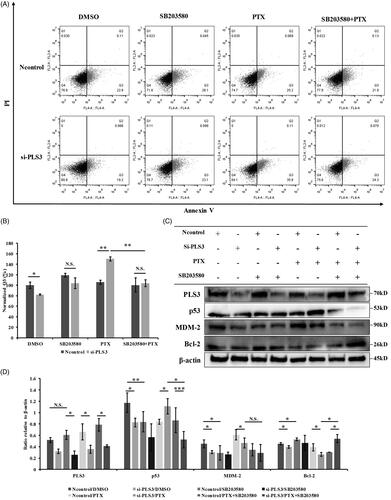
To determine the molecular events responsible for blocking PTX-induced apoptosis by SB203580 in si-PLS3 transfected cells, we analyzed the expression of p53, MDM2 and Bcl-2 by Western blotting in Ncontrol and si-PLS3 transfected cells treated with SB203580 and PTX alone or in combination. As shown in , SB203580 alone decreased the expression of p53, MDM2, and increased the expression of Bcl-2 relative to the Ncontrol/DMSO level in Ncontrol groups, demonstrating that p53, MDM2 and Bcl-2 were involved in p38 MAPK singaling pathway. The results that SB203580 hardly had any effect on their expression in the si-PLS3 group was probably due to si-PLS3 had altered their expressions by inhibiting p38 MAPK already. Among the three proteins, p53 and Bcl-2 were the key effectors responsibe for enhancing PTX-induced apoptosis by si-PLS3 down-regulation, as the combination of PTX with SB203580 only reversed p53 up-regulation and Bcl-2 down-regulation induced by PLS3 down-regulation compared with si-PLS3/PTX (). The results suggested that p53 and Bcl-2, involved in the p38 MAPK signalling pathway, might contribute to the strengthened apoptosis in response to PTX by PLS3 down-regulation.
PLS3 down-regulation may decrease the cell’s sensitivity to ABR by impairing endocytosis
PTX and ABR have different cellular transport mechanisms, where that of PTX is through passive diffusion and that of ABR is through caveolae-mediated endocytosis [Citation27,Citation28]. Thus, we speculated that the cellular uptake mechanism was most likely responsible for the differential apoptotic effects induced by these two PTX drugs. Since PLS3 knockout impaired endocytosis in yeast [Citation29,Citation30], we investigated the role that PLS3 plays in endocytosis in the breast cancer cells.
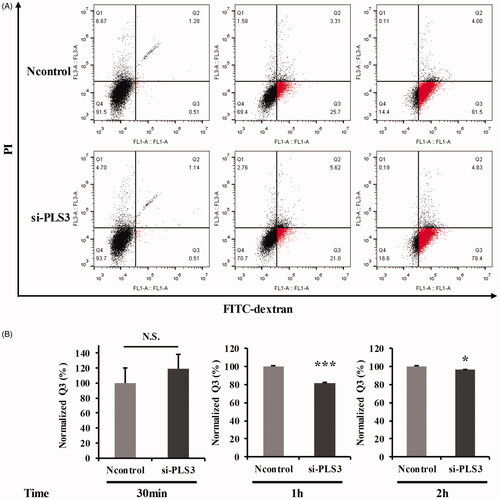
The endocytosis assay was first optimized by subjecting the MDA-MB-231 cells to a low temperature (4 °C), a known endocytosis-disrupting factor. After incubation with FITC-dextran for 1 h, the cellular uptake of FITC-dextran was significantly decreased at 4 °C compared with that at 37 °C (Figure S3). We then analyzed the impact of PLS3 down-regulation on endocytic FITC-dextran uptake in the MDA-MB-231 cells at different time points. The cellular uptake level of FITC-dextran was similar in both the Ncontrol and si-PLS3 groups with treatment for 30 min (). However, after incubation for another 30 min, the endocytotic FITC-dextran uptake was strongly inhibited in the si-PLS3 group. When the incubation time was increased to 2 h, nearly 80% of the si-PLS3-transfected cells had endocytosed FITC-dextran, but there still existed a significant difference in uptake levels between the Ncontrol and si-PLS3 groups. Images captured under a fluorescence microscope (IX73, OLYMPUS, Tokyo, Japan ) at the incubation time of 2 h are shown in Figure S4. These results demonstrated that endocytosis was disturbed or impaired by the PLS3 silencing in the MDA-MB-231 cells and this down-regulation of PLS3 may be the reason for the decreased sensitivity of the cells to ABR.
Figure 5. Down-regulation of plastin 3 (PLS3) may decrease the cell’s sensitivity to Abraxane (ABR) via the impairment of endocytosis. (A) Representative dot plots showing FITC-dextran uptake in cells transfected with Ncontrol or si-PLS3 at 30 min, 1 h, and 2 h. (B) Quantification of the Q3 population and histogram plots showing a decrease in FITC-dextran uptake upon PLS3 down-regulation. The analysis was carried out by using FlowJo 10.0 software. ***p < .001, *p < .05 versus Ncontrol. N.S.: No significance. All the data represent the means ± SD of three independent experiments.
Discussion
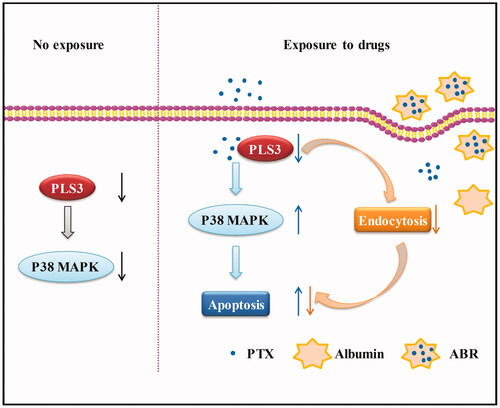
In this study, we characterized the function of PLS3 down-regulation in the sensitivity of breast cancer cells to PTX and ABR. Herein, we postulated that PLS3 down-regulation influences the cancer cell’s sensitivity to PTX and ABR through enhancement of the drug-induced apoptosis. PLS3 down-regulation augments the sensitivity to PTX by enhancing apoptosis via activation of the p38 MAPK signalling pathway, whereas impaired endocytosis neutralizes the apoptosis induced by the free PTX in PLS3-silenced cells exposed to ABR (). Thus, in the present study, we have provided evidence for the effect of PLS3 on the cell’s sensitivity to PTX and ABR and the mechanisms involved.
Figure 6. Schematic model presenting the role of plastin 3 (PLS3) in the regulation of apoptosis induced by paclitaxel (PTX) or Abraxane (ABR) in triple-negative breast cancer cells. In cells with no exposure, PLS3 down-regulation inhibits p38 MAPK signalling pathway. In cells exposed to PTX, PLS3 down-regulation augments the sensitivity to PTX by enhancing apoptosis via activation of the p38 MAPK signalling pathway. In PLS3-silenced cells exposed to ABR, impaired endocytosis neutralises the apoptosis induced by the free PTX.
It is controversial to identify the role of p38 MAPK as a tumour suppressor or promoter on the basis of its capacity to modulate cell survival or apoptosis [Citation31,Citation32]. On the one hand, p38 MAPK acts as a promoter of cell survival. As previously reported, p38 MAPK silencing inhibited the growth of MDA-MB-231 cells in the long term (1 week), whereas it had no effect on the cell growth in 72 h, which is consistent with the result in [Citation33]. We surmised that additional survival signals provided by an activated Ras mutant contributed to the survival of MDA-MB-231 cells when PLS3 down-regulation induced the inhibition of p38 MAPK [Citation34]. On the other hand, p38 MAPK activation mediates apoptosis following various stimuli. In MDA-MB-231 cells, the activation of p38 MAPK was required in the apoptotic response to vorinostat and p38 MAPK silencing could abrogate this effect [Citation35]. Besides this, the suppression of p38 MAPK by SB203580 significantly decreased the apoptosis induced by PTX [Citation9]. In light of our results, the inhibition of p38 MAPK could attenuate the strengthened apoptosis mediated by PLS3 down-regulation in the context of PTX exposure. All of this indicates that p38 MAPK plays a crucial role in the apoptotic response to PTX in MDA-MB-231 cells.
The mechanism of p38 MAPK downstream signalling to mediate apoptosis has long been widely studied but is poorly clarified. Various stimuli result in the activation of p38 MAPK, which then stimulates the complicated downstream network [Citation32]. It is well known that p38 MAPK is involved in the apoptosis induced by many chemical reagents through the regulation of p53 and its downstream effector molecules [Citation36–38]. Herein, we also found that p53 and Bcl-2 were downstream effector molecules in the p38 MAPK-mediated regulation of PTX-induced apoptosis in PLS3-down-regulated cells. Since MDM-2 inhibits the stability of p53, a paradoxical scenario likely existed in our study; namely, that the expression level of p53 did not always correlate with that of MDM-2 ( and ). This scenario may be because p53 degradation depends partially on MDM-2 and there are other ubiquitin ligases responsible for its ubiquitination[Citation39]. Bcl-2, which is negatively regulated by p38 MAPK, also constitutes a mechanism for the induction of apoptosis [Citation40]. Interestingly, the decrease in Bcl-2 expression through PLS3 silencing did not induce apoptosis ( and ), indicating that PLS3 down-regulation may trigger other apoptotic molecules downstream of p38 MAPK aside from Bcl-2. For instance, caspase functions as an activated death protease, catalyzing the specific cleavage of many key cellular proteins and ultimately leading to apoptosis [Citation41]. Herein, we also found a correlation between the decrease in cleaved-caspase 3 and the inhibition of apoptosis mediated by PLS3 down-regulation (Figure S2). Thus, under the circumstance of PLS3 down-regulation, Bcl-2 may not act as an effector molecule for apoptosis suppression since its function may be neutralized by other molecules like caspase-3. Consistent with a previous study, our study also implies that the apoptosis induced by different stimuli depends on different downstream effector molecules via p38 MAPK signalling [Citation31].
ABR is a solvent-free nanoparticle (130 nm) formulation of albumin-bound PTX. It has revealed many advantages over PTX in both preclinical and clinical studies. The differences in clinical efficacy and safety between ABR and PTX are attributed to the special endogenous transport pathways of albumin, resulting in enhanced drug delivery and accumulation in the tumour tissue. Albumin is transported across the endothelial barrier of blood vessels by transcytosis, initiated by its binding to albumin receptor gp60, which in turn results in the binding of gp60 with the intracellular protein caveolin-1 to form caveolae [Citation42–44]. This process may be facilitated through the binding of albumin to secreted protein acid and rich in cysteine (SPARC) [Citation45]. PTX is transported mainly by passive diffusion, whereas ABR is transported by caveolae-mediated endocytosis. Regardless of the different transport pathways, the final cell-killing drug is the free PTX, which targets tubulin. Therefore, the functional distinction in drug sensitivity induced by PLS3 down-regulation is most likely due to the different modes of PTX and ABR transport into the cells. Recent studies reported the function of PLS3 in the regulation of endocytosis, where PLS3 knockout impaired endocytosis in yeast, whereas its overexpression restored the endocytotic level reduced by the deficiency of the SMN gene through direct interaction with the F-actin-binding protein CORO1C in the SMN-deficient cells [Citation28,Citation30]. Moreover, since PLS3 knockdown may impair the expression of F-actin, which is essential in all types of endocytosis, the decrease in endocytosis might be a result of reduced F-actin dynamics [Citation46,Citation47]. With gene therapy development, RNA interference has been considered in combination with chemotherapy [Citation48]. In our study, whereas PLS3 down-regulation strengthened the cell-killing effect of PTX, it failed to perform the same effect with ABR treatment, which limits the efficacy of RNA interference in clinical application. Thus, it is essential for us to consider the form of drugs when studying the effects of combination therapy of PTX and the inhibition of potential targets like PLS3. Collectively, our study suggests that the disruption of endocytosis might be a key cellular mechanism underlying the failure of PLS3 down-regulation to enhance the sensitivity of the MDA-MB-231 cells to ABR, and it is necessary to consider the cellular transport modes for different drugs when studying the effects of certain proteins like PLS3 on drug sensitivity. The precise mechanism by which PLS3 impairs endocytosis needs to be further investigated.
Recently, many research studies on PLS3 have implied its potential role as a marker for cancer prognosis in the clinical setting. For instance, PLS3 was found to be expressed in epithelial-mesenchymal transition (EMT)-induced circulating tumour cells in the peripheral blood of patients with colorectal cancer with distant metastasis, indicating that PLS3 possesses a significant prognostic value [Citation18]. Further research revealed the involvement of PLS3 in the transforming growth factor-beta-induced EMT process [Citation49]. EMT confers resistance to the cell death induced by chemotherapy through various ways in cancer cells, implying a relationship between EMT and chemotherapy-induced apoptosis [Citation50]. Whether the enhancement in cell sensitivity to PTX mediated by PLS3 down-regulation correlates to EMT deserves further study. Our study also suggests that PLS3 could become a potential marker for clinical diagnosis and therapy since PTX conferred enhanced apoptosis in cells with a lower expression of PLS3 compared with that in cells with normal PLS3 expression. This finding indicates that PTX administration may be more effective in patients whose cancer cells express a low amount of PLS3 during the clinical treatment.
In conclusion, we have demonstrated, for the first time, that PLS3 acts as an effector molecule to mediate PTX-induced apoptosis in MDA-MB-231 cells via the p38 MAPK signalling pathway. Furthermore, PLS3 down-regulation failed to confer enhancement of cell sensitivity to the nano-drug ABR, owing to impairment of the endocytotic process triggered by PLS3 down-regulation in the breast cancer cells. Finally, our study not only elucidates the mechanism by which PLS3 regulates the cell’s sensitivity to PTX but also provides a potential therapeutic target and marker for the clinical diagnosis and chemotherapy of TNBC.
Supplemental_file.docx
Download ()Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Goldspiel BR. Clinical overview of the taxanes. Pharmacotherapy. 1997;17:110S–125S.
- Holmes FA, Walters RS, Theriault RL, et al. Phase II trial of taxol, an active drug in the treatment of metastatic breast cancer. J Natl Cancer Inst. 1991;83:1797–1805.
- McGuire WP, Rowinsky EK, Rosenshein NB, et al. Taxol: a unique antineoplastic agent with significant activity in advanced ovarian epithelial neoplasms. Ann Intern Med. 1989;111:273–279.
- Hayes DF, Thor AD, Dressler LG, et al. HER2 and response to paclitaxel in node-positive breast cancer. N Engl J Med. 2007;357:1496–1506.
- Ellis P, Barrett-Lee P, Johnson L, et al. Sequential docetaxel as adjuvant chemotherapy for early breast cancer (TACT): an open-label, phase III, randomised controlled trial. Lancet. 2009;373:1681–1692.
- Hawkins MJ, Soon-Shiong P, Desai N. Protein nanoparticles as drug carriers in clinical medicine. Adv Drug Deliv Rev. 2008;60:876–885.
- Haley B, Frenkel E. Nanoparticles for drug delivery in cancer treatment. Urol Oncol. 2008;26:57–64.
- Subbaramaiah K, Hart JC, Norton L, et al. Microtubule-interfering agents stimulate the transcription of cyclooxygenase-2. Evidence for involvement of ERK1/2 and p38 mitogen-activated protein kinase pathways. J Biol Chem. 2000;275:14838–14845.
- Bacus SS, Gudkov AV, Lowe M, et al. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene. 2001;20:147–155.
- Seidman R, Gitelman I, Sagi O, et al. The role of ERK 1/2 and p38 MAP-kinase pathways in taxol-induced apoptosis in human ovarian carcinoma cells. Exp Cell Res. 2001;268:84–92.
- Lu M, Xiao L, Li Z, et al. The relationship between p38MAPK and apoptosis during paclitaxel resistance of ovarian cancer cells. J Huazhong Univ Sci Technol [Med Sci]. 2007;27:725–728.
- Park SH, Seong MA, Lee HY. p38 MAPK-induced MDM2 degradation confers paclitaxel resistance through p53-mediated regulation of EGFR in human lung cancer cells. Oncotarget. 2016;7:8184–8199.
- Sanchez-Prieto R, Rojas JM, Taya Y, et al. A role for the p38 mitogen-acitvated protein kinase pathway in the transcriptional activation of p53 on genotoxic stress by chemotherapeutic agents. Cancer Res. 2000;60:2464–2472.
- Miyashita T, Krajewski S, Krajewska M, et al. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799–1805.
- George J, Banik NL, Ray SK. Bcl-2 siRNA augments taxol mediated apoptotic death in human glioblastoma U138MG and U251MG cells. Neurochem Res. 2009;34:66–78.
- Bretscher A, Weber K. Fimbrin, a new microfilament-associated protein present in microvilli and other cell surface structures. J Cell Biol. 1980;86:335–340.
- Glenney JR, Jr, Kaulfus P, Matsudaira P, et al. F-actin binding and bundling properties of fimbrin, a major cytoskeletal protein of microvillus core filaments. J Biol Chem 1981;256:9283–9288.
- Yokobori T, Iinuma H, Shimamura T, et al. Plastin3 is a novel marker for circulating tumor cells undergoing the epithelial-mesenchymal transition and is associated with colorectal cancer prognosis. Cancer Res. 2013;73:2059–2069.
- Ueo H, Sugimachi K, Gorges TM, et al. Circulating tumour cell-derived plastin3 is a novel marker for predicting long-term prognosis in patients with breast cancer. Br J Cancer. 2015;112:1519–1526.
- Hisano T, Ono M, Nakayama M, et al. Increased expression of T-plastin gene in cisplatin-resistant human cancer cells: identification by mRNA differential display. FEBS Lett. 1996;397:101–107.
- Ikeda H, Sasaki Y, Kobayashi T, et al. The role of T-fimbrin in the response to DNA damage: silencing of T-fimbrin by small interfering RNA sensitizes human liver cancer cells to DNA-damaging agents. Int J Oncol. 2005;27:933–940.
- Liu H, Ma Y, He HW, et al. SLC9A3R1 stimulates autophagy via BECN1 stabilization in breast cancer cells. Autophagy. 2015;11:2323–2334.
- Zhang H, Zhang S, He H, et al. RasGAP-derived peptide 38GAP potentiates the cytotoxicity of cisplatin through inhibitions of Akt, ERK and NF-κB in colon carcinoma HCT116 cells . Cancer Lett. 2011;308:62–70.
- Giannakakou P, Robey R, Fojo T, et al. Low concentrations of paclitaxel induce cell type-dependent p53, p21 and G1/G2 arrest instead of mitotic arrest: molecular determinants of paclitaxel-induced cytotoxicity. Oncogene. 2001;20:3806–3813.
- Demidenko ZN, Kalurupalle S, Hanko C, et al. Mechanism of G1-like arrest by low concentrations of paclitaxel: next cell cycle p53-dependent arrest with sub G1 DNA content mediated by prolonged mitosis. Oncogene. 2008;27:4402–4410.
- Wang TH, Wang HS, Soong YK. Paclitaxel-induced cell death: where the cell cycle and apoptosis come together. Cancer. 2000;88:2619–2628.
- Yardley DA. nab-Paclitaxel mechanisms of action and delivery. J Control Release. 2013;170:365–372.
- Walle UK, Walle T. Taxol transport by human intestinal epithelial Caco-2 cells. Drug Metab Dispos. 1998;26:343–346.
- Kubler E, Riezman H. Actin and fimbrin are required for the internalization step of endocytosis in yeast. Embo J. 1993;12:2855–2862.
- Hosseinibarkooie S, Peters M, Torres-Benito L, et al. The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am J Hum Genet. 2016;99:647–665.
- Bulavin DV, Fornace AJ. Jr., p38 MAP kinase's emerging role as a tumor suppressor. Adv Cancer Res. 2004;92:95–118.
- Dolado I, Nebreda AR. Regulation of tumorigenesis by p38α MAP kinase. In: Posas F, Nebreda AR, editors. Stress-activated protein kinases. Topics in current genetics. Vol 20. Berlin–Heidelberg (Germany): Springer; 2007. p. 99–128.
- Chen L, Mayer JA, Krisko TI, et al. Inhibition of the p38 kinase suppresses the proliferation of human ER-negative breast cancer cells. Cancer Res. 2009;69:8853–8861.
- Kozma SC, Bogaard ME, Buser K, et al. The human c-Kirsten ras gene is activated by a novel mutation in codon 13 in the breast carcinoma cell line MDA-MB231. Nucleic Acids Res. 1987;15:5963–5971.
- Uehara N, Kanematsu S, Miki H, et al. Requirement of p38 MAPK for a cell-death pathway triggered by vorinostat in MDA-MB-231 human breast cancer cells. Cancer Lett. 2012;315:112–121.
- Kwon YW, Ueda S, Ueno M, et al. Mechanism of p53-dependent apoptosis induced by 3-methylcholanthrene: involvement of p53 phosphorylation and p38 MAPK. J Biol Chem. 2002;277:1837–1844.
- Gao J, Gao J, Qian L, et al. Activation of p38-MAPK by CXCL4/CXCR3 axis contributes to p53-dependent intestinal apoptosis initiated by 5-fluorouracil. Cancer Biol Ther. 2014;15:982–991.
- Lamy V, Bousserouel S, Gosse F, et al. Lupulone triggers p38 MAPK-controlled activation of p53 and of the TRAIL receptor apoptotic pathway in human colon cancer-derived metastatic cells. Oncol Rep. 2011;26:109–114.
- Lukashchuk N, Vousden KH. Ubiquitination and degradation of mutant p53. Mol Cell Biol. 2007;27:8284–8295.
- De CG, Marcocci ME, Torcia M, et al. Bcl-2 Phosphorylation by p38 MAPK: identification of target sites and biologic consequences. J Biol Chem. 2006;281:21353–21361.
- Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104.
- Schnitzer JE. gp60 is an albumin-binding glycoprotein expressed by continuous endothelium involved in albumin transcytosis. Am J Physiol. 1992;262:H246–H254.
- Minshall RD, Tiruppathi C, Vogel SM, et al. Endothelial cell-surface gp60 activates vesicle formation and trafficking via G(i)-coupled Src kinase signaling pathway. J Cell Biol. 2000;150:1057–1070.
- Vogel SM, Minshall RD, Pilipovic M, et al. Albumin uptake and transcytosis in endothelial cells in vivo induced by albumin-binding protein. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1512–L1522.
- Trieu V, Frankel T, Labao E, et al. SPARC expression in breast tumors may correlate to increased tumor distribution of nanoparticle albumin-bound paclitaxel (ABI-007) vs. taxol. Abstract in the Proceedings of the 96th Annual Meeting of the American Association for Cancer Research; 2005 Apr 16–20; Anaheim, CA. Cancer Res. 2005;65(9 Suppl):1314.
- Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902.
- Mooren OL, Galletta BJ, Cooper JA. Roles for actin assembly in endocytosis. Annu Rev Biochem. 2012;81:661–686.
- Pai SI, Lin YY, Macaes B, et al. Prospects of RNA interference therapy for cancer. Gene Ther. 2006;13:464–477.
- Sugimachi K, Yokobori T, Iinuma H, et al. Aberrant expression of plastin-3 via copy number gain induces the epithelial-mesenchymal transition in circulating colorectal cancer cells. Ann Surg Oncol. 2014;21:3680–3690.
- Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751.