
Abstract
Elevated free fatty acids (FFAs) are a risk factor for type 2 diabetes. Endothelial dysfunction induced by high levels of FFAs is one of the mechanisms related to the progression of diabetes. In clinical diabetes care, DPP-4 inhibitors have been shown to be effective in reducing glucose levels. In this study, we investigated the molecular mechanism of the clinically available DPP-4 inhibitor vildagliptin in the protection of FFA-induced endothelial dysfunction. Treatment of endothelial cells with vildagliptin inhibits FFA-induced cellular LDH release and generation of ROS. Vildagliptin also reverses FFA-induced reduced levels of GSH and elevated expression of the FFA-associated NAPHD oxidase protein NOX-4. Moreover, vildagliptin ameliorates the reduction in mitochondrial potential triggered by FFAs. Mechanistically, we show that vildagliptin suppresses FFA-induced expression of proteins of the NLRP3 inflammasome complex, including NLRP3, ASC, p20 and HMGB-1, and mitigates FFA-induced inactivation of the AMPK pathway. Consequently, vildagliptin inhibits production of two cytokines that are favored by NLRP3 inflammasome machinery: IL-1β and IL-18. Finally, we demonstrate that vildagliptin ameliorates FFA-induced reduced eNOS, indicating its protective role against endothelial dysfunction. Collectively, we conclude that the protective role of vildagliptin in endothelial cells is mediated via suppression of the AMPK-NLRP3 inflammasome-HMGB-1 axis pathway. These findings imply that the anti-diabetic drug vildagliptin possesses dual therapeutic applications in lowering glucose and improving vascular function.
Introduction
The epidemic of obesity has become a worldwide health problem in recent decades and nearly 30% of the world’s population was obese or overweight between 1980 and 2013 [Citation1]. Obesity is a disease resulting from a metabolic abnormality and is a major risk factor for both type 2 diabetes and cardiovascular disease [Citation2]. It has been reported that obesity-related type 2 diabetes accounts for more than 90% of all diagnosed cases of diabetes in adults [Citation3]. Although it is well known that cardiovascular complication is the major cause of mortality among diabetes patients, the complex mechanisms driving the impact of diabetes on the heart and vasculature are poorly understood.
Obesity-related type 2 diabetes patients often have elevated plasma FFAs either because an increased amount of FFAs is released from their large amounts of adipose tissue or they have an impaired FFA clearance mechanism [Citation4]. Elevated plasma levels of FFAs disturb insulin signaling and often cause a whole-body imbalance in metabolism. Hyperinsulinemia related to abnormal FFA metabolism is a direct risk factor for atherosclerosis as well as other cardiovascular disorders. Elevated plasma levels of FFAs also cause a high degree of inflammation, which also contributes to cardiovascular disease.
Endothelial cells line the inside walls of the vascular system and serve as an interface between circulating blood and the surrounding tissue. Owing to its high density, elevated plasma levels of FFAs could directly disturb the endothelial NO production mechanism, which is dependent on endothelial PI3K-eNOS signaling. Thus, endothelial cells often have reduced eNOS activity and diminished NO production in diabetic conditions [Citation5]. FFAs also contribute to higher production of ROS and inflammation in endothelial cells [Citation6].
Many drug discoveries have entered into clinical practice to improve vascular complications in type 2 diabetes. DPP-4 inhibitors are effective agents for lowering blood glucose in type 2 diabetes patients. More than a dozen DPP-4 inhibitor agents have been developed for the treatment of type 2 diabetes, including the most commonly used alogliptin, linagliptin, saxagliptin, sitagliptin, and vildagliptin. In recent years, several different DPP-4 inhibitors have been shown to be effective in improving endothelial function, reducing oxidative and pro-inflammatory states, and exerting beneficial effects on cardiovascular function [Citation7–9]. In our current study, we investigated the molecular mechanism of vildagliptin in cultured human endothelial cells.
Materials and methods
Cell culture and treatment experiment
Primary human umbilical vascular endothelial cells (HUVECs) were purchased from Lonza (Basel, Switzerland) (CC-2517). HUVECs were grown in 2% serum endothelial growth medium (EGM2) in low passage numbers (less than 10). All cell cultures were maintained in 5% (v/v) CO2/95% (v/v) nitrogen incubator at 37 °C. FFAs were prepared with oleic and linoleic acids (Sigma-Aldrich, St Louis, USA). Briefly, oleic and linoleic acids were dissolved in 0.1 M NaOH at 70 °C and then complexed with 10% BSA at 55 °C for 10 min. These stock solutions of 5 mM FFAs were prepared the day before the experiment, placed in a sealed tube in a nitrogen atmosphere and stored overnight at -20 °C, then added to the cell culture medium. A solution with 10% BSA was added to control cultures. For FFA treatment, 1 mM prepared FFAs [Citation10] containing growth media was added to confluent HUVECs media for 6, 24, or 48 h, respectively, depending on the experiment. For vildagliptin treatment experiments, 2.5 and 5 μM vildagliptin was added to confluent HUVECs media for 24 h.
Quantitative PCR analysis
The total RNA from HUVECs was extracted using a micro RNeasy Micro Kit from Qiagen (Duesseldorf. German) (Cat.74004) in accordance with the manufacturer’s manual. The RNA concentration was quantified by Nanodrop. A total of 1 μg of RNA was used to synthesize cDNA using iScript™ Reverse Transcription Supermix (BioRad, Hercules, USA, #1708840) for RT-qPCR from Invitrogen (Carlsbad, USA). For quantitative PCR, SYBR-based real-time PCR experiments were performed to detect the total mRNA transcripts of human IL-1β, IL-18, eNOS, NOX-4, HMG-1, and GAPDH using an ABI 7500 platform.
Western blot analysis
HUVECs under the different conditions were lyzed by RIPA buffer with protease and phosphatase inhibitors. The nuclear extracts were obtained by lysing the cells with hypotonic buffer to remove the cytoplasm. Cell lysates or nuclear extracts (20 μg) were immobilized by PAGE gel. The separated protein mixes were then transferred onto PVDF membranes and blotted against their specific antibodies and corresponding secondary antibody. The immunoblots were visualized by Pierce™ ECL Plus western blotting substrate (Promega, Madison, USA) (Catalog # 32132).
Lactate dehydrogenase (LDH) release
Cytotoxicity of the HUVECs was assessed by measuring leakage of lactate dehydrogenase (LDH) into the culture medium. The culture medium was collected to obtain cell-free supernatants from the different conditions. The activity of LDH in the media was determined using a commercially available kit from Thermo Fisher Scientific, Waltham, USA.
ROS and GSH/GSSG assays
Cellular production of reactive oxygen species (ROS) was measured by quick staining the cells under the different conditions with 2′, 7′–dichlorofluorescein diacetate dye (DCFDA). The fluorescent images were captured using a fluorescence microscope. The fluorescent density of the images was quantified using Image J software (NIH, Bethesda, USA).
To measure cellular oxidative stress, we quantified the ratio of the reduction in glutathione (GSH) level. We purchased the kit from Thermo Fisher Scientific, USA. The assay was performed by following the manufacturer’s protocol.
Mitochondrial membrane potential assay
Mitochondrial membrane potential (MMP) assay was based on the measurement of TMRM staining (tetramethylrhodamine methyl and ethyl esters). Based on the concentration of TREM accumulation in the mitochondria, the aggregated fluorescent counts were measured to determine mitochondrial depolarization and cell health status. We purchased a TREM kit (Thermo Fisher Scientific, USA) and measured MMP activity, with fluorescent density and quantified to present the data.
Statistical analysis
Data are shown as means ± standard derivation (SD). All two-group comparisons in our study were tested using the Student’s two-tailed t test. All three-group comparisons were performed using the one-way analysis of variance (ANOVA) test, and p values less than .05 was determined to be statistically significant.
Results
Vildagliptin protects endothelial cells against cytotoxicity induced by high free fatty acids (FFAs)
To investigate the role of vildagliptin in endothelial cells, we challenged HUVECs with high free fatty acids (FFAs) and then tested the influence of vildagliptin on FFA-induced cellular toxicity by measuring the release of lactate dehydrogenase (LDH). As shown in , non-treated HUVECs had only about 5% LDH release and the addition of 1 mM FFAs caused LDH to release to rise to approximately 47%. However, FFAs caused only 25% and 15% LDH release in the presence of 2.5 and 5 μM vildagliptin, respectively, suggesting that vildagliptin could have a protective role by reducing FFAs-induced LDH release.
Figure 1. Vildagliptin inhibits FFA-induced endothelial lactate dehydrogenase (LDH) release. Human umbilical vein endothelial cells (HUVECs) were treated with high FFAs (1 mM) in the presence or absence of vildagliptin (2.5, 5 μM) for 48 h. LDH release was determined using a commercial kit (a, b, c, p < .01 vs. previous column group).
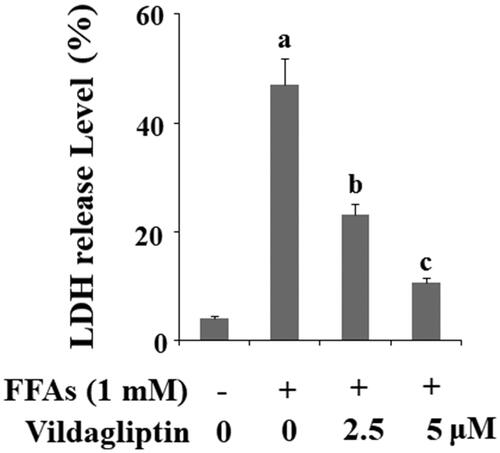
Vildagliptin inhibits FFA-induced generation of reactive oxygen species (ROS)
Next, we measured cellular production of ROS under the various conditions. As shown in the representative images in , there are very low-level ROS production under basal non-treated conditions and the presence of FFAs significantly increased the generation of ROS. However, the addition of vildagliptin appears to have a weakening effect. When these images were normalized and presented as a plot, we concluded that FFAs caused approximately 3.5-fold higher ROS production as compared with the control, whereas FFAs caused only approximately 2- and 1.3-fold higher ROS in the presence of the two doses of vildagliptin, respectively. We then measured the reduction in GSH level, an important indicator of intracellular oxidative stress. As shown in and compared to the control, FFAs caused a reduction in GSH by half. However, the two doses of vildagliptin gradually recovered approximately 70 and 90% of the normal GSH level. Clearly, vildagliptin has an inhibitory role on FFA-induced production of ROS and could retain the level of GSH in HUVECs.
Figure 2. Vildagliptin suppresses FFA-induced endothelial generation of reactive oxygen species (ROS) and reduction of glutathione (GSH). (A). Intracellular ROS was determined by DCFH-DA; (B). Intracellular GSH levels. Human umbilical vein endothelial cells (HUVECs) were treated with high FFAs (1 mM) in the presence or absence of vildagliptin (2.5, 5 μM) for 48 h (a, b, c, p < .01 vs. previous column group).
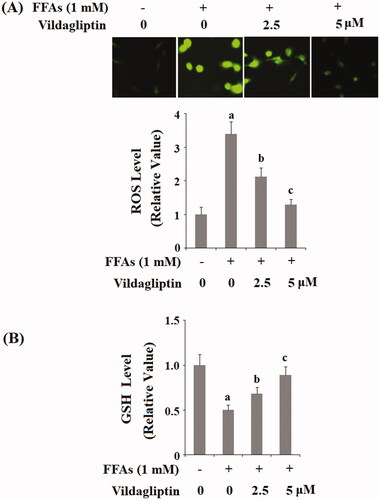
Vildagliptin suppresses FFA-induced endothelial NOX-4 expression
The NADPH oxidase complex contributes to cellular ROS production and NOX-4 is its major subunit expressed in the endothelial cells. We assessed the endothelial level of NOX-4 in these circumstances. shows that at the mRNA transcript level, FFAs strongly induced approximately 6-fold higher NOX-4 expression as compared to the control. However, FFAs gave rise to only approximately 3.8- and 2-fold expression of NOX-4 in the presence of the two doses of vildagliptin, respectively. This inhibition of NOX-4 was also verified at the protein level. As presented in and compared to the control, FFAs induced 3.5-fold NOX-4 protein expression, but only approximately 2.3 and 1.5-fold NOX-4 expression in the presence of the same two doses of vildagliptin, respectively.
Figure 3. Vildagliptin suppresses FFA-mediated endothelial NOX-4 elevation. Human umbilical vein endothelial cells (HUVECs) were treated with high FFAs (1 mM) in the presence or absence of vildagliptin (2.5, 5 μM) for 24 h. (A). mRNA levels of NOX-4 as determined by real-time PCR; (B). Protein levels of NOX-4 determined by western blot analysis (a, b, c, p < .01 vs. previous column group).
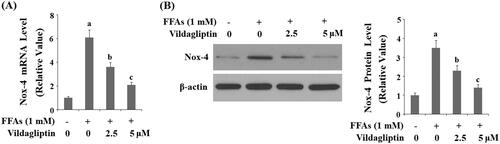
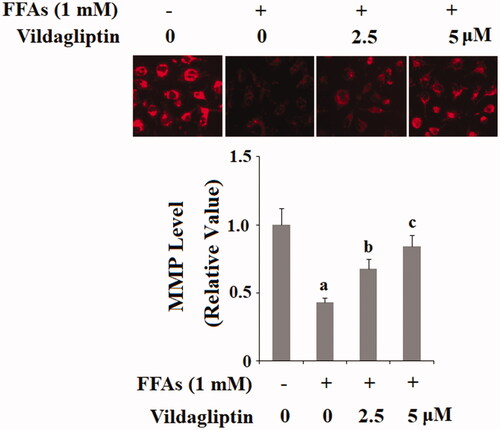
Vildagliptin protects against FFA-induced endothelial mitochondrial function
Another source of ROS production is an endothelial mitochondrial malfunction. We reasoned that the anti-ROS property of vildagliptin could be attributed to its protective effect on endothelial mitochondrial function. By measuring mitochondrial membrane potential (MMP), we assessed alterations in mitochondrial function under the different conditions. As shown in , there is a fair amount of mitochondrial membrane staining under basal conditions, while exposure to FFAs caused a reduction in membrane staining by about half. However, the addition of the two doses of vildagliptin restored the MMP level by approximately 70 and 85% in the presence of the same concentration of FFAs. These results implicate that vildagliptin protects FFA-induced loss of endothelial mitochondrial function.
Figure 4. Vildagliptin restores FFA-induced reduced endothelial mitochondrial membrane potential (MMP). Human umbilical vein endothelial cells (HUVECs) were treated with high FFAs (1 mM) in the presence or absence of vildagliptin (2.5 μM, 5 μM) for 48 h. MMP levels were determined by TMRM staining (a, b, c, p < .01 vs. previous column group).
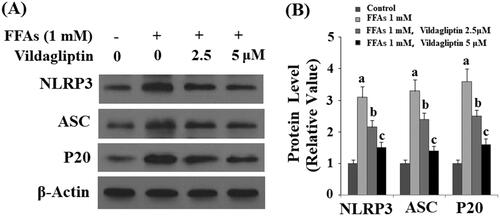
Vildagliptin prevents FFA-induced NLRP3 inflammasome activation in endothelial cells
Endothelial cells are a direct target of blood cell-derived inflammatory mediators and other pathogens, called DAMPs and PAMPs (damage- or pathogen-associated molecular pattern molecules). The inflammasome complex has been identified as a major source of numerous cytokines in the DAMP- or PAMP-mediated immune response [Citation11]. We tested the influence of vildagliptin on the most well-characterized endothelial inflammasome NLRP3 complex. As shown by the results of western blot analysis in and the quantified plot in (normalized to non-treated cells), FFA stimulation gave rise to 3- to 4-fold higher expression of all three components of the NLRP3 complex including NLRP3, ASC, and p20 protein. However, the presence of 2.5 and 5 μM vildagliptin ubiquitously restored the expression of all three proteins to normal levels in a dose-dependent manner. These data indicate that vildagliptin suppresses activation of the inflammasome NLRP3 complex in endothelial cells.
Figure 5. Vildagliptin prevents FFA-induced endothelial NLRP3 inflammasome activation. Human umbilical vein endothelial cells (HUVECs) were treated with high FFAs (1 mM) in the presence or absence of vildagliptin (2.5, 5 μM) for 24 h. (A). Western blot analysis of NLRP3, ASC, and cleaved caspase 1 (P20); (B). Quantification of western blot results (a, b, c, p < .01 vs. previous column group).
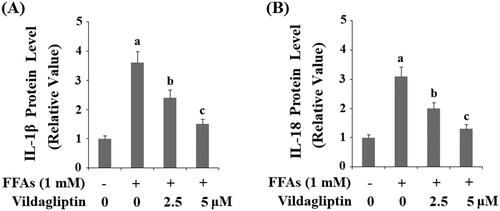
Vildagliptin inhibits FFA-induced production of IL-1β and IL-18
Next, we measured cytokines levels in the presence of this combined treatment. As shown in and compared to the control, FFAs caused approximately 3.5-fold higher IL-1β protein production. However, FFAs gave rise to only approximately 2.5- and 1.5-fold higher IL-1β in the presence of the two doses of vildagliptin, respectively. Vildagliptin has a similar suppressive effect on the production of another cytokine, IL-18. As shown in and compared to the control, FFAs gave rise to approximately 3-fold higher IL-18 expression. However there was only approximately 2- and 1.5-fold IL-18 production in the presence of the two doses of vildagliptin, respectively.
Figure 6. Vildagliptin inhibits FFA-induced maturation of IL-1β and IL-18. (A). Secretion of IL-1β was assessed by ELISA; (B). Secretion of IL-18 was assessed by ELISA. Human umbilical vein endothelial cells (HUVECs) were treated with high FFAs (1 mM) in the presence or absence of vildagliptin (2.5, 5 μM) for 48 h (a, b, c, p < .01 vs. previous column group).
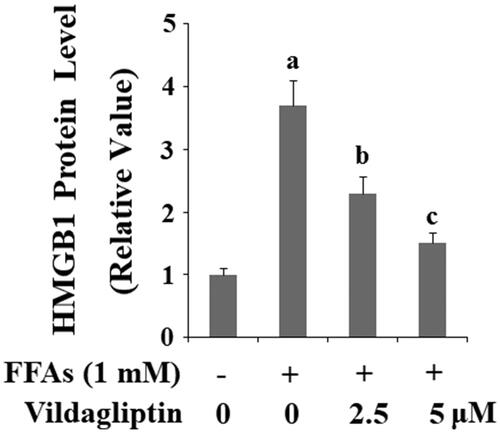
Vildagliptin inhibits FFA-induced endothelial-derived HMGB-1 expression
High mobility group box 1 protein (HMGB-1) has been shown to be the responsive chromatin protein involved in inflammasome activation in endothelial cells (Fiuza C, 2003). We measured the levels of endothelial-derived HMGB-1 in a separate treatment experiment. As shown in and compared to the control, FFA treatment resulted in approximately 3.6-fold higher expression of HMGB-1 protein. However, FFAs gave rise to only approximately 2.4- and 1.5-fold higher expression of HMGB-1 in the presence of the two doses of vildagliptin, respectively.
Figure 7. Vildagliptin inhibits FFA-induced release of high mobility group box-1 protein (HMGB1). Human umbilical vein endothelial cells (HUVECs) were treated with high FFAs (1 mM) in the presence or absence of vildagliptin (2.5 μM, 5 μM) for 48 h. Secretion of HMGB1 was determined by ELISA (a, b, c, p < .01 vs. previous column group).
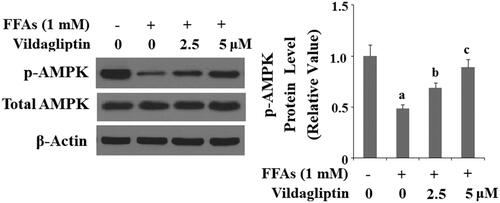
Vildagliptin mitigates FFA-mediated deactivation of AMPK
AMPK activation is vital to many aspects of endothelial function and has been shown to be the upstream stress response kinase responsible for producing numerous cytokines. Therefore, we assessed the influence of vildagliptin on activation of AMPK by phosphorylated AMPK. As shown in and compared to the control, FFA treatment resulted in a reduction of p-AMPK by about half. However, the addition of the two doses of vildagliptin preserved approximately 75 and 95% of the basal level of p-AMPK, despite the action of FFAs, respectively.
Figure 8. Vildagliptin mitigates FFA-induced AMPK inactivation. Human umbilical vein endothelial cells (HUVECs) were treated with high FFAs (1 mM) in the presence or absence of vildagliptin (2.5 μM, 5 μM) for 6 h. Phosphorylated and total AMPK was determined by western blot analysis (a, b, c, p<.01 vs. previous column group).
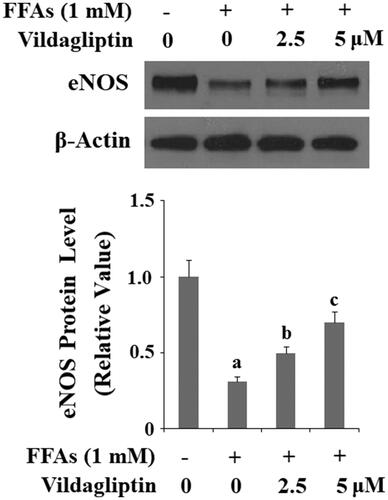
Vildagliptin ameliorates FFA-mediated eNOS reduction
Finally, we assessed the effect of vildagliptin on endothelial function by measuring eNOS protein levels. As shown in and compared to the control, treatment of HUVECs with FFAs diminished eNOS expression by 70%. However, the same concentration of FFAs decreased eNOS expression by only approximately 50 and 30% in the presence of the two doses of vildagliptin, respectively. Therefore, we conclude that vildagliptin has a protective effect on endothelial function based on its influence on eNOS level.
Figure 9. Vildagliptin suppresses FFA-induced reduction of eNOS. Human umbilical vein endothelial cells (HUVECs) were treated with high FFAs (1 mM) in the presence or absence of vildagliptin (2.5 μM, 5 μM) for 24 h. eNOS was determined by Western blot analysis (a, b, c, p < .01 vs. previous column group).
Discussion
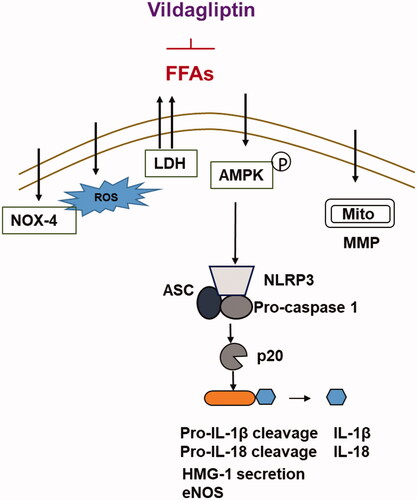
Several members of the DPP-4 inhibitor family of medications have been shown to exert vascular effects independent of their glucose-lowering ability [Citation12]. Vildagliptin is a member of the DPP-4 family. In light of this, we investigated the molecular basis of its action in cultured endothelial cells. Our data demonstrate that vildagliptin exerts significant cellular protective effects under insult by high FFAs in cultured endothelial cells. FFA-induced production of mitochondrial ROS plays an important role in the inflammatory response and subsequent endothelial dysfunction [Citation13]. NLRP3-dependent pyroptosis and maturation of pro-inflammatory cytokines, such as IL-1β and IL-18, induced by ROS have been linked with the pathological progression of cardiovascular diseases [Citation14]. FFA treatment induces mitochondrial dysfunction accompanied by reduced mitochondrial membrane potential (MMP) in endothelial cells [Citation15]. NOX-4 is an important mitochondrial enzyme, which mediates ROS production. Importantly, increased NOX-4 expression is associated with NLRP3 activation and IL-1β maturation in endothelial cells [Citation16]. Importantly, blockage of NOX-4 activation with its specific inhibitor can prevent NLRP3 inflammasome activation and reduce caspase-1 cleavage as well as the production of IL-1β and IL-18 [Citation17]. Elevation of LDH activity is an important consequence of NLRP3-ASC inflammasome activation, pyroptosis, and production of IL-1β and IL-18 [Citation18]. Here, we present five facts as evidence of the protective roles of vildagliptin in the context of a high FFA environment: (1). The detoxifying capacity of vildagliptin reduces the endothelial release of LDH. Our data indicate that the action of vildagliptin is potent enough to nearly abolish FFA-induced release of LDH. This finding prompted our investigation into all aspects of its protective effects. (2). The anti-ROS property of vildagliptin can be reflected by our data showing that vildagliptin reduces cellular ROS levels and recovers reduced cellular GSH levels resulting from FFA-induced oxidative stress. In our experiment, vildagliptin strongly inhibited NOX-4 induction, suggesting that the capacity of vildagliptin to suppress ROS production could be mediated by its action on endothelial NOX-4. (3). The anti-inflammatory property of vildagliptin is evidenced by its suppression of the production of inflammatory cytokines (IL-1β and IL-18), the release of chromatin protein HMGB-1 and NLRP3 inflammasome. The NLRP3 inflammasome has been shown to be crucial in controlling the maturation of the two pro-inflammatory cytokines IL-1β and IL-18 [Citation19]. The NLRP3 complex is activated upon binding of DAMPs and PAMPs (damage or pathogen-associated molecular pattern molecules). DAMPs and PAMPs are released by the immune system in response to tissue injury and pathogen invasion. DAMPs and PAMPs activate the functional NLRP3 inflammasome by initiating assembly of a multi-protein complex consisting of NLRP3, the adaptor protein ASC, and pro-caspase-1. The consequence of activation of the NLRL3 complex is the production of the cleaved form of caspase-1 (p20) [Citation20]. Caspase-1 is known as an inflammatory caspase that plays a role in the maturation of IL-1β and IL-18 into active cytokines. In our experiment, vildagliptin appears to inhibit all of the major components of the NLRP3 inflammasome complex, including NLRP3, ASC, and p20. Secretion of HMGB1 has been shown to be a downstream effect of NRLR3 inflammasome assembly and caspase 1 activation [Citation21]. The suppression of HMGB-1 by vildagliptin further confirmed its action on NLRP3 inflammasome. Thus, we conclude that vildagliptin is a strong suppressor of the NLRP3 inflammasome. (4). The suppression of NLRP3 inflammasome by vildagliptin is mediated by its suppression of AMPK signaling. Our data show that vildagliptin suppresses endothelial AMPK inactivation and recent studies have linked the AMPK pathway as a modulator of NLRP3 inflammasome activation [Citation22,Citation23]. Therefore, the inhibitory role of vildagliptin on inflammation could be mediated by its impact on AMPK signaling. (5). Vildagliptin is a beneficial factor for protecting endothelial function. A graphic summary of the underlying mechanism is shown in . We demonstrate that vildagliptin greatly recovers eNOS level, suggesting that vildagliptin could play an important role in maintaining the vasoreactivity of endothelial cells.
Figure 10. Graphic summary of the underlying mechanisms.
Other studies have documented that vildagliptin plays various roles in vascular function. Preclinical animal studies have shown that administration of vildagliptin could improve vascular integrity and protect against diabetic condition-induced vascular senescence in diabetic rats [Citation24,Citation25]. Another study showed that vildagliptin treatment enhances blood flow recovery and capillary density in ischemic animals [Citation26]. In a vascular dementia rat model, administration of vildagliptin significantly improved blood-brain barrier function [Citation27]. A recently published one-year randomized controlled clinical trial indicated that vildagliptin administration mitigates the reduction in the number of endothelial progenitor cells in type 2 diabetes patients [Citation28]. Another recent study showed that vildagliptin treatment protects endothelial cells from loss of identity towards transformation into other cell types and improves pulmonary fibrosis in mice [Citation29]. All these evidences demonstrate that vildagliptin possesses an array of vascular protective effects. However, the molecular mechanism of vildagliptin in improving endothelial function remains largely elusive. Our study adopted the physiologically relevant method of challenging endothelial cells with high FFAs and examined the detailed molecular mechanism through which vildagliptin exerts its effects. The AMPK-NLRP3 inflammasome axis regulated by vildagliptin is the highlight of the mechanism.
The main limitation of the current study is that we only examined the protective effects of vildagliptin against FFA-induced NLRP3 inflammasome activation and the inflammatory response in an in vitro primary endothelial culture model. It should be noted that the pathological mechanisms of endothelial dysfunction are complicated and need to be further elucidated. Diverse risk factors, including genetics, aging, and obesity, are reported to be involved in cardiovascular disease [Citation30]. Animal experimentation is an important method for exploring the pathogenesis and treatment of cardiovascular diseases. In addition, the pathogenesis of cardiovascular diseases is regulated through a complex network of signaling pathways. It is possible that vildagliptin plays a role in regulating the crosstalk of signaling pathways involved in FFA-induced endothelial dysfunction. Future investigations with animal models or clinical trials are necessary to verify the pharmacological function of vildagliptin in vivo. Vascular endothelial dysfunction is present in diabetes and its vascular complications. Now, cardiovascular diseases have emerged as a common complication of diabetes and improved strategies for the prevention and treatment of cardiovascular diseases are evolving rapidly. Dysfunction of vascular cells has been shown to contribute to the pathogenesis and clinical expression of atherosclerosis in diabetes. Elucidation of the molecular mechanism behind the protective effects of vildagliptin on vascular function will provide valuable data for its implication in the treatment of diabetes and its cardiovascular complications.
Disclosure statement
None of the authors of this article have any conflicts of interest that require disclosure.
References
- Ng M, Fleming T, Robinson M, The GBD 2013 Obesity Collaboration. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384:766–781.
- Scherer PE, Hill JA. Obesity, diabetes, and cardiovascular diseases: a compendium. Circ Res. 2016;118:1703–1705.
- Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics–2015 update: a report from the American Heart Association. Circulation. 2015;131:e29–322.
- Boden G. Effects of free fatty acids (FFA) on glucose metabolism: significance for insulin resistance and type 2 diabetes. Exp Clin Endocrinol Diabetes. 2003;111:121–124.
- Dresner A, Laurent D, Marcucci M, et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999;103:253–259.
- Inoguchi T, Li P, Umeda F, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–1945.
- Avogaro A, Kreutzenberg S, Fadini G. Dipeptidyl-peptidase 4 inhibition: linking metabolic control to cardiovascular protection. CPD. 2014;20:2387–2394.
- Lei Y, Hu L, Yang G, et al. Dipeptidyl peptidase-IV inhibition for the treatment of cardiovascular disease – recent insights focusing on angiogenesis and neovascularization. Circ J. 2017;81:770–776.
- Nauck MA, Meier JJ, Cavender MA, et al. Cardiovascular actions and clinical outcomes with glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Circulation. 2017;136:849–870.
- Xue Y, Guo T, Zou L, et al. Evodiamine attenuates P2X7-mediated inflammatory injury of human umbilical vein endothelial cells exposed to high free fatty acids. Oxid Med Cell Longev. 2018;2018:1.
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426.
- Fiuza C, Bustin M, Talwar S, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–2660.
- Jaimes EA, Hua P, Tian RX, et al. Human glomerular endothelium: interplay among glucose, free fatty acids, angiotensin II, and oxidative stress. Am J Physiol Renal Physiol. 2010;298:F125–F132.
- Liu D, Zeng X, Li X, et al. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res Cardiol. 2018;113:5.
- Knapp A, Czech U, Góralska J, et al. Influence of fatty acids on mitochondrial metabolism of adipocyte progenitors and endothelial cells. Arch Physiol Biochem. 2012;118:128–134.
- Qi J, Yu XJ, Shi XL, et al. NF-κB blockade in hypothalamic paraventricular nucleus inhibits high-salt-induced hypertension through NLRP3 and caspase-1. Cardiovasc Toxicol. 2016;16:345–354.
- Moon JS, Nakahira K, Chung KP, et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat Med. 2016;22:1002–1012.
- Wu X, Zhang H, Qi W, et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018;9:171.
- Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735.
- Jo EK, Kim JK, Shin DM, et al. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. 2016;13:148–159.
- Lamkanfi M, Sarkar A, Vande Walle L, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185:4385–4392.
- Lv H, Liu Q, Wen Z, et al. Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/GSK3β-Nrf2 signal axis. Redox Biol. 2017;12:311–324.
- Cordero MD, Williams MR, Ryffel B. AMP-activated protein kinase regulation of the NLRP3 inflammasome during aging. Trends Endocrinol Metab. 2018;29:8–17.
- Wang D, Luo P, Wang Y, et al. Glucagon-like peptide-1 protects against cardiac microvascular injury in diabetes via a cAMP/PKA/Rho-dependent mechanism. Diabetes. 2013;62:1697–1708.
- Oeseburg H, de Boer RA, Buikema H, et al. Glucagon-like peptide 1 prevents reactive oxygen species-induced endothelial cell senescence through the activation of protein kinase A. ATVB. 2010;30:1407–1414.
- Ishii M, Shibata R, Kondo K, et al. Vildagliptin stimulates endothelial cell network formation and ischemia-induced revascularization via an endothelial nitric-oxide synthase-dependent mechanism. J Biol Chem. 2014;289:27235–27245.
- Jain S, Sharma B. Neuroprotective effect of selective DPP-4 inhibitor in experimental vascular dementia. Physiol Behav. 2015;152:182–193.
- Dei Cas A, Spigoni V, Cito M, et al. Vildagliptin, but not glibenclamide, increases circulating endothelial progenitor cell number: a 12-month randomized controlled trial in patients with type 2 diabetes. Cardiovasc Diabetol. 2017;16:27.
- Suzuki T, Tada Y, Gladson S, et al. Vildagliptin ameliorates pulmonary fibrosis in lipopolysaccharide induced lung injury by inhibiting endothelial-to-mesenchymal transition. Respir Res. 2017;18:177.
- Zhaolin Z, Guohua L, Shiyuan W, et al. Role of pyroptosis in cardiovascular disease. Cell Prolif. 2018;e12563.