
Abstract
This study aimed to induce spermatogenesis in azoospermic testis through induced pluripotent stem cells (iPSCs) derived spermatogonial stem cell-like cells (SSCLCs) after iPSCs in vivo and in vitro transplantation and three-dimensional organ culture. DiI-labelled mouse iPSCs were transplanted to azoospermic testis mouse model (pretreated by busulfan 40 mg/kg). This study was designed based on two experimental groups. In experimental group 1(in vivo) labelled iPSCs were transplanted to azoospermic host testis. In experimental group 2 (in vitro) after cell transplantation, fragments of host testes were set as 3D organ culture and testis without cells transplantation served as the control group by the same method. The samples were evaluated by tracing DiI, cell homing, immunohistochemistry, and quantitative RT PCR assays. 2 weeks after iPSCs transplantation, the molecular assessment showed that Plzf, Thy1, Vasa and Gfra1 expression were increased significantly (p ≤ .05) in host testis and labelled iPSCs co-localized by the Plzf and Thy1 markers expression in the base of seminiferous tubules. These findings suggest the ability of iPSCs to achieve homing in the testis niche and indicate the critical inductive role of microenvironment signals in the differentiation of iPSCs to spermatogonial stem cell-like cells.
Introduction
Infertility is a global public health problem with a high incidence in couples of reproductive age [Citation1]. According to the World Health Organization, ∼15% of the couples in the world are infertile, of which 50% are due to male factors. Male infertility generally affects 7% of the male population, also 10% of infertile men are azoospermic [Citation2]. Azoospermia occurs due to genetic factors, injuries, exposure to toxins, immune-suppressive and anticancer therapies [Citation3]. Recently, advanced techniques such as testicular sperm extraction (TESE) and intra cytoplasmic sperm injection (ICSI) have been applied for male infertility treatments. Unfortunately, these techniques have been unable to help infertile men who lack functional sperm. Generation of germ cells and induction of spermatogenesis from pluripotent stem cells may have a great potential for treating infertility [Citation4]. Derivation of autologous germ cells for cell therapy and injecting them back into the patient to regenerate damaged tissues without the need to repress the immune system is one of the benefits of using iPSCs in stem cell therapy, tissue engineering, reproductive and regenerative medicine [Citation5]. iPSCs are pluripotent and therefore have the ability to differentiate into various types of cells if they are placed in a suitable niche. Based on previous studies, male germ cells induction from iPSCs has rapidly advanced since 2009. Park et al. reported the first attempt to create primordial germ cell-like cells (PGCLCs) from human iPSCs by co-culturing with human fetal gonadal stromal cells [Citation6]. In this field, similar researches have been carried out in a variety of different protocols, for example, addition of cytokines and induction factors such as bone morphogenetic protein 4 (BMP4), retinoic acid (RA) to culture media, genetic manipulation and the other methods [Citation7–11]. But there is no perfect in vitro protocol for iPSCs differentiation toward male germ cells. It should be noted that the in vitro differentiation of iPSCs to germ cells needs culture conditions that are similar to that of spermatogonial stem cells (SSCs) niche in the testis, in order to achieve more advanced, efficient and effective derivation of male germ cells from iPSCs. To evaluate the effect of testicular niches on iPSCs differentiation, we transplanted iPSCs directly into cell-depleted seminiferous tubules of the mouse. We also employed in vitro transplantation and (3D) testicular organ culture that was established by Yokonishi to obtain spermatogonial stem cell-like cells (SSCLCs) from iPSCs [Citation12]. We hope that in vitro culture systems of iPSCs will open a novel path to reproductive engineering and male infertility research. Here, we have demonstrated that testicular organ culture following iPSCs in vitro transplantation can be a suitable and desirable approach to induce initiation of spermatogenesis.
Method and materials
Animals
This research was approved by the Ethics Committee of Tarbiat Modares University, Tehran, Iran. All male Naval Medical Research Institute (NMRI) mice were 6–8 weeks old. These mice were kept in 12-h light-dark cycle at a stable temperature (22 ± 2 °C) and specific pathogen-free environment with easy access to food and water.
Design of study
This study was designed based on two experimental groups. Animals were randomly divided into two groups each containing five mice. In experimental group 1 (in vivo), right testis received iPSCs through efferent duct and other testis set as the control group. In experimental group 2 (in vitro), after cells injection, fragments of host testes were set as 3D organ culture and testis without cell transplantation served as the control group of the same method.
Induction of mouse azoospermic model
To generate the experimental azoospermic model, mice were treated with busulfan (Sigma, St Louis, MO, USA) at 40 mg/kg body weight. The mice azoospermia model was ready 4 weeks after injection [Citation13]. To confirm the azoospermia model, busulfan treated animal was sacrificed by cervical dislocation, the testis was removed from the body and fixed in Bouin’s solution for 48 h, dehydrated and embedded in paraffin. The hematoxylin-eosin (H&E) staining was used for testis assessment in mouse azoospermia model to prove the absence of spermatogenesis.
Preparation and characterization of mouse iPSCs
Mouse iPSC line was donated by Prof. Soleimani (Bonyakhte Stem Cells Technology Research Center, Tehran, Iran). iPSCs were established from male NMRI mouse fibroblasts through retroviral transfer of transcription factors Oct4/Sox2/Klf4/c-Myc. Immunocytochemistry staining by pluripotency markers was performed for their assessment. iPSCs in culture were fixed with 4% formaldehyde for 30 min and rinsed by Phosphate-buffered saline (PBS) twice, then fixed cells were permeabilized with 0.3% Triton X-100 in PBS for 30 min and blocked with 10% goat serum (Sigma-Aldrich, Germany) in PBS. The cells were subjected to specific immunostaining overnight at 4 °C by using the following primary antibodies: anti-Oct4, anti-Klf4 and anti Sox2, all from Abcam UK. The dilution for all of the primary antibodies was 1:100. For immunofluorescence assays FITC-conjugated secondary antibodies (Sigma-Aldrich, Germany) were used at 1:200 in PBS for 60 min at 37 °C in darkness. The secondary antibody was completely removed from cells and was washed by PBS. iPSCs were mounted by 4′, 6-diamidino-2-phenylindole dihydrochloride (DAPI) (Calbiochem, Nottingham, UK) and analyzed by fluorescent microscopy (Olympus, type CH2).
Mouse iPSCs culture and labelling
For the preparation of mouse iPSCs culture, 1.2 g high glucose Dulbecco’s modified Eagle’s medium (Gibco, life technologies, UK) and 0.24 g of sodium bicarbonate (Sigma-Aldrich, St Louis, MO, USA), were solved in 100 ml of double distilled water (DDW) and was sterilized by 0.22-µm Millipore membrane filter. The medium was supplemented with 15% Knock out serum replacement (KOSR), 0.1 mM non-essential amino acids, 1 mM L-glutamine, 0.1 mM mercaptoethanol, 1 × penicillin-streptomycin (all from Gibco, Life Technologies.UK), 1000U/ml leukemia inhibitory factor (LIF) (Millipore, USA). The iPSCs were cultured on a feeder layer of inactivated mouse embryonic fibroblasts (MEFs) in gelatinized dishes. After 2 days of culture, the clones of iPSCs formed on MEFs. Then cells were passaged in the medium every 3 to 5 days and the medium was changed every day (). Before cell transplantation, iPSCs were labelled with Dil (Invitrogen, Cergy Pontoise, France) following the manufacturer’s instructions. Briefly, iPSCs were incubated in PBS containing Dil (4 μg/ml final concentration) for 20 min at 4 °C. (). After that PBS was removed from labelled cells, they were trypsinized and dispersed and re-suspended in culture medium at a concentration of 1 × 105 μl for cells transplantation.
Figure 1. Mouse iPSCs colonies. Morphology of mouse iPSCs colonies after 2 days of culture on a layer of inactivated MEFs.
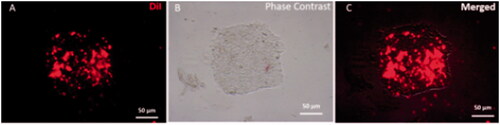
Figure 2. DiI labelled iPSCs colony Fluorescence image of iPSCs colony that labelled by DiI (A) Phase-contrast microscopy of iPSCs colony morphology. (B) Merged image of iPSCs and DiI labelled colony (C).
Agarose gel stands and organ culture plate preparation
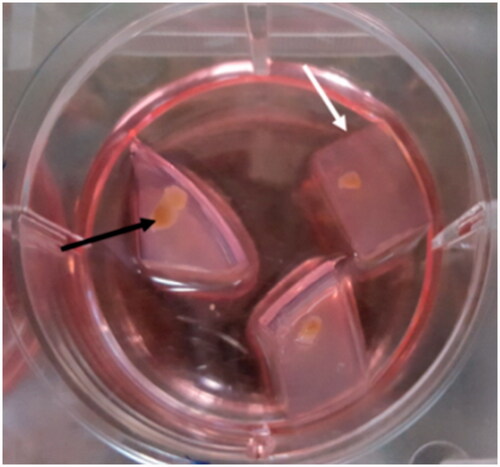
We prepared agarose gel stands by dissolving 1.5 g of agarose (Agar, Carl Roth GmbH + Co. KG) in 100 ml of DDW and autoclaved it. In each 6-cm sterile dish, 11 ml of agarose solution was poured and was left to cool under the laminar flow hood. The gel was cut into pieces of 10 mm × 10 mm × 5 mm. Three or four pieces of agarose were placed in each well of 6 wells plate and the gel was soaked in culture medium for 24 h before use at 34 °C in a CO2 incubator to replace water ( white arrow). For the preparation of organ culture medium, 1.01 g α- minimum essential medium (α-MEM) and 0.22 g of sodium bicarbonate were dissolved in 100 ml of double distilled water (DDW). Subsequently, 1 ml of penicillin-streptomycin was added to medium and then filter-sterilized through a 0.22 µm Millipore membrane filter and stored in the refrigerator (4 °C). Organ culture medium was supplemented by 10% KOSR, progesterone 60 ng/ml (Sigma .UK), LIF 10 ng/ml, glial cell line-derived neurotrophic factor (GDNF) 10 ng/ml (Royan Biotech. Iran), β-estradiol 30 ng/ml (Sigma. UK), recombinant human epidermal growth factor (EGF) 20 ng/ml (Peprptech .UK), and basic fibroblast growth factor (BFGF) 10 ng/ml (Royan Biotech. Iran).
Figure 3. Preparation of agarose gel stand and 3D organ culture. The prepared gel agarose was cut into pieces of 10 mm × 10 mm × 5 mm in size. Three or four pieces of agarose gel were placed in each well of 6 wells plate and were soaked with the medium. The testis was fragmented into ∼1 cm × 1 cm × 1 cm pieces to be appropriate for organ culture. Testes fragments were put on the agarose gel in the three-dimensional organ culture method. Agarose gel is shown by the white arrow, and fragmented testis is pointed by the black arrow.
Transplantation of iPSCs to mouse azoospermic testis
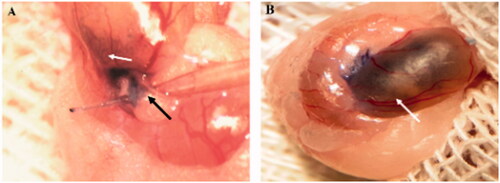
The host testis of the experimental groups received approximately 105 of cultured iPSCs in 10 μl media into the seminiferous tubules. Cell transplantation was done by introducing a microinjection needle into the efferent ducts toward seminiferous tubules (, black arrow). To ensure the cells entering the seminiferous tubules Trypan blue was added to the injection media. ∼70–90% of the tubules were filled in each recipient testis (, white arrow). For magnification and better visibility, cells transplantation was performed under the stereomicroscope (Olympus SZ60).
Figure 4. Cells transplantation into seminiferous tubules. The cell suspension was injected into seminiferous tubules through the efferent duct using a fine microinjection needle (A). ∼70–90% of seminiferous tubules were filled in each recipient testis. (B) Seminiferous tubules are shown by the white arrow, and the efferent duct is indicated by black arrow.
In experimental group 1, azoospermic mouse was anesthetized with 10% ketamine (Rotexmedica, Trittau, Germany) and 20% xylazine (Alfasan, Woerden, Netherlands). Skin of the scrotal area was shaved and disinfected by 10% povidone iodine solution. Right testis was accessed through the scrotal incision and received labelled iPSCs, then host testis was put in the scrotum and the incision sutured. All operations were performed under sterile condition.
In experimental group 2 the mouse was sacrificed by cervical dislocation and testis and epididymis were removed from the body and labelled iPSCs were transplanted as described. Host testis was encapsulated and divided into 18–20 pieces of 1.5–2 mm diameter with sterile fine scissors under the laminar flow hood. Small fragments of host testis were placed on each agarose gel in the prepared culture plate (, black arrow) and then were set under 3D organ culture condition. The medium level was below the upper edge of the agarose gel so that tissue could continue growing without getting mixed-up or becoming suspended in the culture medium. The culture incubator was supplied by 5% CO2 in the air and the temperature maintained at 34 °C. The medium was changed twice a week by replacing the old medium with the same volume of fresh culture medium. Samples were examined by invert microscope (LABOMED, TCM 400, Los Angeles, CA, USA). In both experimental groups, all assessments were performed 2 weeks after cells transplantation and organ culture.
Histopathological evaluation
For histological study, the testes of mice in all groups of our study were fixed in a Bouin’s solution for 48 h, embedded in paraffin, sectioned at 5-μm thickness, and finally stained with hematoxylin-eosin (H&E) for light microscopic visualization.
Real-time PCR studies
Expression of promyelocytic leukaemiaz finger (Plzf), mouse vasa homologous (vasa or Mvh), thymocyte differentiation antigen 1 (Thy1) and GDNF family receptor alpha-1 (Gfra1) as spermatogonial stem cells specific markers was evaluated by real-time PCR. Total RNA was extracted from experimental testis after 2 weeks using Trizol. Genomic contamination was eliminated by treating the RNA with DNase I (Fermentas, Germany). The concentration of RNA was measured by UV spectrophotometry (Eppendorf, Germany). The cDNAs synthesis was done using RevertAid™ First Strand cDNA Synthesis kit (Fermentas, Germany) and oligo (dT) primers. For PCR reactions and for the RT reaction to evaluate target genes and β-actin expression gene-specific primers were designed by the NCBI website. (http://www.ncbi.nlm.nih.gov/) and synthesized by a commercial source (CinnaGen, Iran). Primer sequences and products size are listed in . The PCRs were done using Master Mix and SYBR Green I by Applied Biosystems, StepOne™ thermal cycler (Applied Biosystems, USA). The PCR program was started by a melting cycle of 5 min at 95 °C. This stage was followed by 40 cycles of melting (30 s at 95 °C), annealing (30 s at 60 °C), and extension (30 s at 72 °C). The quality of the PCR reactions was confirmed by the melting curve analyses. A standard curve was used to determine the gene efficiency (logarithmic dilution series of cDNA from the testes). The target genes and the reference (β-actin) were amplified in the same run. In addition, this process was repeated and duplicated three times for all the target and reference genes. Expression assay of stage-specific marker for Plzf, Vasa, Thy1 and Gfra1 genes was analyzed using the comparative ΔΔCT method. The target genes were normalized against the reference gene. Then the target genes’ expressions in experimental groups were investigated in relation to the genes expressions in iPSCs before transplantation.
Table 1. Primers used for qRT PCR analysis of spermatogonial stem cells gene markers.
Immunostaining assessment
For immunohistochemical staining, the sections were deparaffinized and rehydrated, then washed in PBS solution for 5 min. The sections were placed in Sodium citrate buffer at 100 °C for 20–30 min. Subsequently, they were washed with PBS three times, for 5 min. To block non-specific binding sites, after incubation with 0.3% Triton-X100, slides were also treated with a blocking solution containing 10% goat serum in PBS for 1 h at 37 °C. For detection of Plzf and Thy1markers, the sections were treated with monoclonal antibody against Plzf (1:100, Sigma Aldrich, Germany) and Thy1(1:100, Sigma Aldrich, Germany) left at 4 °C overnight. After it was washed by PBS, the second antibody FITC (1:200, Sigma-Aldrich, Germany) was applied at 37 °C for 2 h in darkness. Then the section was rinsed by PBS, the nuclei were counterstained by DAPI at 0.1 μg/ml for 5 min, and the slides were washed in PBS. Transplanted iPSCs in the seminiferous tubules of recipient testes were detected by tracing DiI labelled cell on the same tissue sections using the fluorescent microscope (Olympus, type CH2).
Statistical analysis
Gene expression analysis was performed using one-way analysis of variance and Tukey post-test using Graph Pad Prism 5.0 (Graph Pad Software, San Diego, CA). The p-values <0.05 was considered statistically significant. All data were shown as mean ± SD from at last three independent experiments.
Results
Characterization of mouse iPSCs
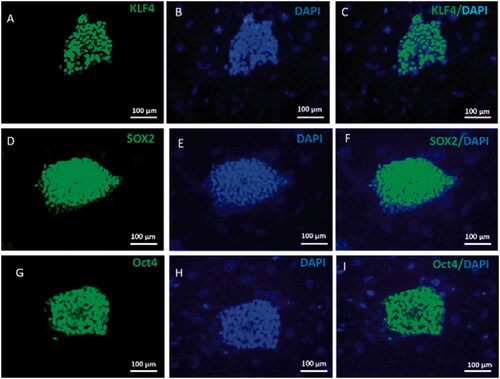
In this study, verification of mouse iPSCs pluripotency was done through immunostaining for Sox2, Oct4 and Klf4 markers. Results are shown in indicating strong positive of mouse iPSCs for these markers while inactivated mouse embryonic fibroblast was negative ().
Figure 5. Mouse iPSCs Characterization. Mouse iPSCs were characterized by immunofluorescence staining and specific pluripotent antibodies including Oct4, Sox2 and Klf4. iPSCs were strongly positive for pluripotent stem cell markers Oct-4, Sox2 and Klf4 (green) (A, D, G). Nuclei were stained blue with DAPI. (B, E, H). The merged picture of iPSCs colony with positive pluripotent stem cell markers and DAPI staining (C, F, I).
Organ culture morphological study
Samples were evaluated by an inverted microscope during 2 weeks of organ culture. The changes such as darkness at the center that may be an indication of necrosis or apoptosis were not observed and seminiferous tubules were lucent ().
Figure 6. Photomicrograph of tissue sections following 2-weeks culture. In Photomicrograph of tissue sections following 2-weeks culture; the clearness in the center of the tissue section represents the tubules access to nutrition.

Histological assessment
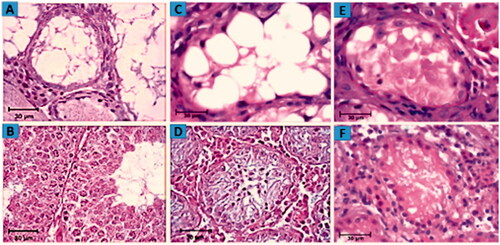
Histological analysis of the testes in mouse azoospermia model revealed that 4 weeks after busulfan treatment, most seminiferous tubules contained no apparent spermatid and spermatozoa. These tubules contained mostly a single basal row of Sertoli cells nuclei and spermatogonial stem cells. In addition, Leydig cells were present in the interstitial space ( and ). Assessment of morphological changes in seminiferous tubules in experimental group 1 showed some tubule repopulation from stem cells and demonstrated little improvement ( and ). In experimental group 2, mouse testes were cut into small pieces, so seminiferous tubules were ruptured and got cut off. Therefore, in the histological section, they are not regularly visible like other normal testicular sections and ). The results after 2 weeks of culture indicated that in experimental group 2, seminiferous tubules had gained rebuilding and restoring spermatogenesis ( and ). Despite the improvement in the epithelium of seminiferous tubule structure following iPSCs transplantation, some deformed and depleted seminiferous tubules were still observed in the testes following 2 weeks after organ culture.
Figure 7. Assessment of morphological changes. H&E staining showing the morphology of mouse testes after 4 weeks of 40 mg/kg busulfan treatment most seminiferous tubules are depleted and no spermatogenetic activity. (A) Wild type mouse testis without busulfan treatment, seminiferous tubules contain spermatogonia, spermatid and spermatozoa. (B) Host testis 2 weeks after iPSCs transplantation and morphological changes from in vivo studies. Restoration of spermatogenesis in seminiferous tubule following iPSCs transplantation is illustrated (C) compared to the control group (D). Histological section of testis fragment cultured for 2 weeks following iPSCs transplantation and 3D organ culture. Due to the cutting of seminiferous tubules, they are not seen regularly in the tissue section. The initiation of spermatogenesis process is visible in the seminiferous tubule of host testis fragments (E) in comparison to control testis (F).
Molecular assessment
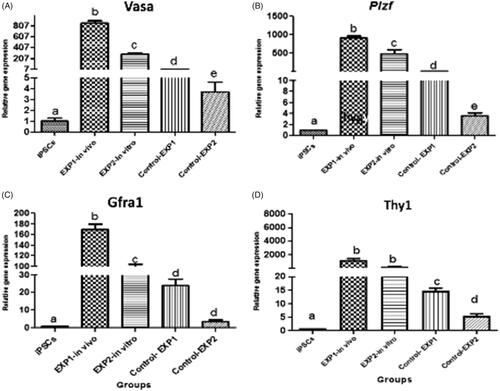
According to the results, iPSCs transplantation in mice significantly increased the expression of premiotic genes compared with controls. The expression of Plzf, Vasa, Thy1 and Gfra1 genes, 2 weeks after in vivo transplantation of iPSCs to azoospermic mice testis increased significantly when compared with the control group (). The above-mentioned markers in cultured tissue were greater than its control group counterpart (p ≤ .05). In control groups, little expression of target genes were also observed. Based on the data, target genes expression were undetected in iPSCs ().
Figure 8. Relative gene expression profile of pre-meiotic Plzf, Vasa, Thy1 and Gfra1 by qRT-PCR. Gene expression of target genes in iPSCs before and 2 weeks after cells transplantation in experimental groups were evaluated by Real Time PCR (qPCR). Data were normalized againt β-actin and represented by mean ± SE after three times repeats. Target gene expressions in experimental groups were investigated in relation to genes expression in iPSCs before transplantation. There are Different letters indicating statistically significant differences between groups (p < .05).
Cells tracing analysis and immunohistochemical results
Fluorescent red light indicated that before transplantation, iPSCs colonies had been labelled with DiI (). 2 weeks after transplantation, fluorescent-labelled cells were followed up in the recipient testes by fluorescent red light. Review of images showed that the DiI positive cells are localized in the base of seminiferous tubules and they showed homing of the transplanted cells ( and ).
Figure 9. Representative immunohistostaining of SSCs markers in testis section 2 weeks after in vivo cells transplantation and 3D organ culture DiI fluorescent-labelled cells were traced in the recipient testes by fluorescent red light. Images (A, E, I and M) show that the DiI positive cells are localized at the base of seminiferous tubules and have gained homing. The host testis sections were immunostained by the antibodies for Plzf and Thy1 (green) (B, F, J and N). DAPI was used for nuclei staining. (C, G, K and O). The co-localization Plzf and Thy1with DiI labelled iPSCs (D, H, L and P) around seminiferous tubules in recipient testes transplanted is shown by fluorescent microscope.
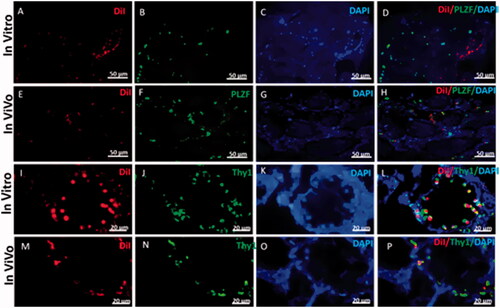
Plzf and Thy1 are specific markers of spermatogonial stem cells. 2 weeks after transplantation, fluorescent-labelled cells were localized at the base of seminiferous tubules of the recipient testes ( and ).
We witnessed Plzf and Thy1 positive cells at the base of tubules that co-localized with DiI-labelled cells in both in vivo and in vitro experimental groups ( and ).
Discussion
Although male germ cells induction from iPSCs has rapidly developed since 2009, transplantation and clinical use of autologous male germ cells induced from iPSCs to restore spermatogenesis in infertile men still require considerable caution. In this study, we transplanted mouse iPSCs to seminiferous tubules of azoospermic mouse model without any genetic or laboratory manipulation. Our results showed that 2 weeks after iPSCs in vitro transplantation and 3D organ culture and in vivo transplantation, transplanted cells migrated to the base of seminiferous tubules, achieved homing and spermatogenesis process was started. In molecular studies, relative Plzf, Vasa, Thy1 and Gfra1 expression in host testis is greater than that of the control group. Also, immunohistochemical studies and tracing DiI-labelled cells in host testis showed expression of pre-meiotic Plzf and Thy1or CD90 markers in labelled cells. Plzf (Zfp145) is a transcriptional repressor protein and is expressed in SSCs and undifferentiated spermatogonia [Citation14–16]. This gene is a known spermatogonial specific marker which adjusts the epigenetic state of undifferentiated spermatogonia and is essential for spermatogonial stem cell maintenance in an undifferentiated state and progenitor cells [Citation14,Citation17]. In rodents, bovine, primates and goat undifferentiated spermatogonia and SSCs express surface marker of THY1 [Citation18,Citation19]. Vasa also known as Mvh is a particular marker for differentiating germ cells from late migration to post-meiotic stage. It is also considered as a dependable marker for differentiation of germ cells from stem cells [Citation19]. Gfra1 is a cell surface receptor and is expressed in undifferentiated spermatogonia. It is a co-receptor for GDNF, that controls the equilibrium between self-renewal and differentiation of SSCs [Citation20]. According to the results of previous studies and morphological and molecular data in the present research, following the injection of the iPSCs into the seminiferous tubules and their exposure to the testicular microenvironments, they were differentiated into male germ cells [Citation9,Citation10]. Also, most studies have proved stem cells differentiate into mature and functional cells based on signals from the microenvironment [Citation21–23]. Therefore, these changes in the transplanted iPSCs may be due to the effects of the testis niche and microenvironment. The niche is a complex of tissue cells, extracellular matrix, substrates, and chemical and physical factors, and one or more stem cells types have homing there. The niche can regulate self-renewal, progeny production and differentiation of stem cells [Citation21–23]. The testis environment and SSCs niches significance was discovered in 1994 by Brinster et al. [Citation24]. Also, they found that transplantation of spermatogonial stem cells to seminiferous could cause spermatogenesis restoration [Citation25].The SSCs niche includes the basic structural and basement membrane of the seminiferous tubule, Sertoli cell, peritubular myoid cells and Leydig cells. In this study, azoospermia model was created 4 weeks following busulfan injection [Citation13]. Histological analysis of testis indicated that more tubules were devoid of any germ cells and contained mostly a single, basal row of Sertoli cells nuclei. In fact, in this condition the Sertoli cells were naked and available, thus the vacant spaces between Sertoli cells serve as hosts for transplanted iPSCs and provided a suitable niche for cells homing. Sertoli cells or testis nurse cells are polarized epithelial cells and provide a microenvironment that is very important and essential for nutritional support, proliferation and differentiation of germ cells [Citation26,Citation27]. They interact with one another through a unique complex termed as ectoplasmic specializations [Citation27,Citation28]. Cytokine factors and proteins secreted by the Sertoli cells are necessary for development, maintenance and self-renewal of spermatogonial stem cells. GDNF is one of the essential proteins that is produced and secreted by Sertoli cells and regulated by follicle stimulating hormone (FSH) signaling pathway [Citation17,Citation27]. GDNF has the critical role of SSC renewal, differentiation, the proliferation of undifferentiated spermatogonia, and expression of some important genes for maintenance of self-renewing SSCs [Citation29–31]. Stem cell factor (SCF) is another Sertoli cell product and is necessary for germ cells to develop beyond type A spermatogonial stages [Citation17]. Leydig cells are present in clusters in the interstitial space and produce testosterone in response to stimulation by luteinizing hormone (LH) [Citation32]. Testosterone is a major and important androgen in the testis that regulates spermatogenesis [Citation32]. The peritubular myoid cells produce colony-stimulating factor 1 (CSF1) for SSCs maintenance [Citation29]. This complex of mechanical and chemical factors may turn off pluripotency genes or turn on and activate the expression of differentiation genes. In the opinion of some scholars, development of germ cells is accompanied by initiation of DNA demethylation, therefore, the seminiferous tubule microenvironment may cause iPSCS to undergo DNA demethylation, which leads to start of spermatogenesis [Citation10,Citation33]. Although, in 3D organ culture, iPCS took advantage of testis niche to differentiate, and also culture medium was supplemented by the growth factors and cytokines that involved in proliferation and differentiation of germ cells, but the expression of target genes was decreased in this group compared with experimental group 1. One of the important differences between in vivo and in vitro conditions would be the presence of micro-circulatory systems that are obligatory for sustaining the homeostatic function of every organ and tissue. It should be noted that rupture and deformity of seminiferous tubules in 3D organ culture with the unbalanced composition of Leydig and peritubular myoid cells and lack of hypothalamus–pituitary–gonadal axis (HPG) can reduce the efficiency of 3D organ culture compared with in vivo transplantation [Citation34,Citation35]. The 3D testicular culture method has many advantages which can be attributed to the observation of transplanted cells details, the possibility of studying the stages of spermatogenesis progression and study of the cells homing at any time. Also, the use of this method can avoid the occurrence of tumorigenic, immunological and ethical problems. Recently, the neonatal mouse testicular tissue was cultured by this technique for 12 weeks. Histological studies, and the specific genes expression of the germinal cells studies indicated that the expression of germ-cells specific genes elevated after 12 weeks in cultured tissue and the process of spermatogenesis was initiated [Citation36]. actually, this culture technique is some kind of in vitro culture of the testicular natural microenvironment, creating a suitable niche for homing and differentiation of transplanted cells. Our molecular assessment results have shown few male germ cells genes expression in control groups, which may be due to in-situ negligible numbers of spermatogonial stem cells in azoospermic testicular tissue.
Conclusion
Our results show that we have induced spermatogonial stem cells-like cells differentiation from iPSCs following in vitro transplantation and 3D testicular organ culture and in vivo transplantation. This study was done without any artificial manipulation or using high-risk induction factors. In vitro culture is a system mimicking native testis microenvironment for SSCs colonization and can be used to explore mechanisms controlling the differentiation of male germ cells from iPSCs. The method and results of this study are new ideas for male infertility research, has the potential for applications in the treatment of male infertility and reproduction of rare (at risk of extinction) animals. By applying modifications and improving culture conditions, we hope to achieve better results in the future.
Acknowledgements
All experiments have been performed the by Faculty of Medical Science, Tarbiat Modares University, Tehran, Iran. We are grateful to Mr. S. Pourbeiranvand and Ms. Z. Khosrowpour directors of Anatomy laboratory for their helpful and valuable guidance. The Authors greatly appreciate Bonyakhte Stem Cells Technology Research Center for the donation of iPSCs line.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Inhorn MC, Patrizio P. Infertility around the globe: new thinking on gender, reproductive technologies and global movements in the 21st century. Hum Reprod Update. 2015;21:411–426.
- Miyamoto T, Minase G, Okabe K, et al. Male infertility and its genetic causes. J Obstet Gynaecol Res. 2015;41:1501–1505.
- Skakkebaek NE, Rajpert-De Meyts E, Main KM. Testicular dysgenesis syndrome: an increasingly common developmental disorder with environmental aspects: opinion. Hum Reprod. 2001;16:972–978.
- Vloeberghs V, Verheyen G, Haentjens P, et al. How successful is TESE-ICSI in couples with non-obstructive azoospermia? Hum Reprod. 2015;30:1790–1796.
- Fang F, Li Z, Zhao Q, et al. Human induced pluripotent stem cells and male infertility: an overview of current progress and perspectives. Hum Reprod. 2018;33:188–195.
- Park TS, Galic Z, Conway AE, et al. Derivation of primordial germ cells from human embryonic and induced pluripotent stem cells is significantly improved by coculture with human fetal gonadal cells. Stem Cells. 2009;27:783–795.
- Panula S, Medrano JV, Kee K, et al. Human germ cell differentiation from fetal-and adult-derived induced pluripotent stem cells. Hum Mol Genet. 2011;20:752–762.
- Eguizabal C, Montserrat N, Vassena R, et al. Complete meiosis from human induced pluripotent stem cells. Stem Cells. 2011;29:1186–1195.
- Durruthy Durruthy J, Ramathal C, Sukhwani M, et al. Fate of induced pluripotent stem cells following transplantation to murine seminiferous tubules. Hum Mol Genet. 2014;23:3071–3084.
- Ramathal C, Durruthy Durruthy J, Sukhwani M, et al. Fate of iPSCs derived from azoospermic and fertile men following xenotransplantation to murine seminiferous tubules. Cell Reports 2014;7:1284–1297.
- Hayashi K, Ohta H, Kurimoto K, et al. Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 2011;146:519–532.
- Yokonishi T, Sato T, Katagiri K, et al. In vitro spermatogenesis using an organ culture technique. In: Carrell D, Aston K, editors. Spermatogenesis. Totowa, NJ: Humana Press; 2013. p. 479–488.
- Anjamrooz SH, Movahedin M, Mowla SJ, et al. Assessment of morphological and functional changes in the mouse testis and epididymal sperms following busulfan treatment. Iranian Biomed J. 2007;11:15–22.
- Pieri N, Souza A, Mançanares A, et al. Immunolocalization of proteins in the spermatogenesis process of canine. Reprod Dom Anim. 2017;52:170–176.
- Grisanti L, Falciatori I, Grasso M, et al. Identification of spermatogonial stem cell subsets by morphological analysis and prospective isolation. Stem Cells 2009;27:3043–3052.
- Costoya JA, Hobbs RM, Barna M, et al. Essential role of Plzf in maintenance of spermatogonial stem cells. Nat Genet. 2004;36:653.
- Dadoune J-P. New insights into male gametogenesis: what about the spermatogonial stem cell niche? Folia Histochem Cytobiol. 2007;45:141–147.
- Abbasi H, Tahmoorespur M, Hosseini SM, et al. THY1 as a reliable marker for enrichment of undifferentiated spermatogonia in the goat. Theriogenology 2013;80:923–932.
- Reding SC, Stepnoski AL, Cloninger EW, et al. THY1 is a conserved marker of undifferentiated spermatogonia in the pre-pubertal bull testis. Reproduction 2010;139:893–903.
- Grasso M, Fuso A, Dovere L, et al. Distribution of GFRA1-expressing spermatogonia in adult mouse testis. Reproduction 2012;143:325.
- Eshghi S, Schaffer DV. Engineering microenvironments to control stem cell fate and function. In: Melton G, editor. StemBook. Amsterdam, Netherlands: IOS Press; 2008.
- Lane SW, Williams DA, Watt FM. Modulating the stem cell niche for tissue regeneration. Nat Biotechnol. 2014;32:795.
- Reinwald Y, Bratt J, El Haj A. Pluripotent stem cells and their dynamic niche. In: Tomizawa M, editor. Pluripotent stem cells: from the bench to the clinic. London, UK: InTech; 2016.
- Brinster RL, Zimmermann JW. Spermatogenesis following male germ-cell transplantation. Proc Nat Acad Sci. 1994;91:11298–11302.
- Brinster RL, Avarbock MR. Germline transmission of donor haplotype following spermatogonial transplantation. Proc Nat Acad Sci. 1994;91:11303–11307.
- Shlush E, Maghen L, Swanson S, et al. In vitro generation of Sertoli-like and haploid spermatid-like cells from human umbilical cord perivascular cells. Stem Cell Res Ther. 2017;8:37.
- Griswold MD. 50 years of spermatogenesis: Sertoli cells and their interactions with germ cells. Biol Reprod. 2018;99:87–100.
- Fu C, Rojas T, Chin AC, et al. Multiple aspects of male germ cell development and interactions with Sertoli cells require inositol hexakisphosphate kinase-1. Scientific Rep. 2018;8:7039.
- Phillips BT, Gassei K, Orwig KE. Spermatogonial stem cell regulation and spermatogenesis. Phil Trans R Soc B. 2010;365:1663–1678.
- Oatley JM, Avarbock MR, Brinster RL. Glial cell line-derived neurotrophic factor regulation of genes essential for self-renewal of mouse spermatogonial stem cells is dependent on Src family kinase signaling. J Biol Chem. 2007;282:25842–25851.
- Oatley JM, Avarbock MR, Telaranta AI, et al. Identifying genes important for spermatogonial stem cell self-renewal and survival. Proc Nat Acad Sci. 2006;103:9524–9529.
- Zirkin BR, Papadopoulos V. Leydig cells: formation, function and regulation. Biol Reprod. 2018;99:101–111.
- Grabole N, Tischler J, Hackett JA, et al. Prdm14 promotes germline fate and naive pluripotency by repressing FGF signalling and DNA methylation. EMBO Rep. 2013;14:629–637.
- Chimento A, Sirianni R, Casaburi I, et al. Role of estrogen receptors and G protein-coupled estrogen receptor in regulation of hypothalamus–pituitary–testis axis and spermatogenesis. Front Endocrinol. 2014;5:1.
- Sato T, Katagiri K, Yokonishi T, et al. In vitro production of fertile sperm from murine spermatogonial stem cell lines. Nat Commun. 2011;2:472.
- Alrahel A, Movahedin M, Mazaheri Z, et al. Study of Tnp1, Tekt1, and Plzf genes expression during an in vitro three-dimensional neonatal male mice testis culture. IBJ. 2018;22:258–263.