
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Nanoparticles (NPs) have gained importance in addressing drug delivery challenges across biological barriers. Here, we reformulated pentamidine, a drug used to treat Human African Trypanosomiasis (HAT) in polymer based nanoparticles and liposomes and compared their capability to enhance pentamidine penetration across blood brain barrier (BBB). Size, polydispersity index, zeta potential, morphology, pentamidine loading and drug release profiles were determined by various methods. Cytotoxicity was tested against the immortalized mouse brain endothelioma cells over 96 h. Moreover, cells monolayer integrity and transportation ability were examined for 24 h. Pentamidine-loaded polycaprolactone (PCL) nanoparticles had a mean size of 267.58, PDI of 0.25 and zeta potential of –28.1 mV and pentamidine-loaded liposomes had a mean size of 119.61 nm, PDI of 0.25 and zeta potential 11.78. Pentamidine loading was 0.16 µg/mg (w/w) and 0.17 µg/mg (w/w) in PCL NPs and liposomes respectively. PCL nanoparticles and liposomes released 12.13% and 22.21% of pentamidine respectively after 24 h. Liposomes transported 87% of the dose, PCL NPs 66% of the dose and free pentamidine penetration was 63% of the dose. These results suggest that liposomes are comparatively promising nanocarriers for transportation of pentamidine across BBB.
Introduction
Human African Trypanosomiasis (HAT) is a fatal vector borne trypanosomal infection characterized by wasting condition in which there is a slow progressive loss of condition accompanied by increasing anaemia and weakness to the point of extreme emaciation, collapse and death often due to heart failure [Citation1,Citation2]. The disease is endemic in 36 countries of Sub-Saharan Africa and impacts about 70 million people [Citation3–5]. Two distinct subspecies of the Trypanosoma brucei are the causative parasites of the disease [Citation6]. Trypanosoma brucei gambiense which is confined to the Central and West Africa causes a chronic infection and Trypanosoma brucei rhodesience which is found in the East and South of Africa causes an acute form of the disease [Citation3].
The disease occurs in two stages and control relies on both therapeutics and vector control. In the first stage (haemo-lymphatic), pentamidine isethionate is the first line drug of choice for treating T.b. gambiense and Suramin for treating T.b. rhodesiense [Citation7]. In the second stage, (neurological) melarsoprol is the main choice for the treatment of both T.b. gambiense and T.b. rhodesiense infections; Eflornithine against T.b. gambiense; Nifurtimox against T.b. gambiense; and eflornithine in combination with nifurtimox against T.b. gambiense [Citation8,Citation9].
The major problems and challenges in treatment of HAT are the level of toxicities exhibited by the drugs [Citation1,Citation10]. Pentamidine isethionate (see chemical structure in ) is an aromatic dicationic diamidine molecule [Citation11,Citation12] which is a white crystalline powder soluble in water and glycerine and insoluble in acetone, ether and chloroform. It is chemically designated as 4, 4′ diamidino-dephenoxypentane di (β hydroxyethanesulfonate) [Citation13]. It is administered daily by deep intramuscular administration at a dose of 4 mg/kg for 7–10 days [Citation8,Citation12,Citation14]. Patients treated with pentamidine are reported to experience undesirable side effects including abdominal pain, diarrhoea, nausea, vomiting [Citation15] cardiac arrhythmias, very high or low blood sugar, pancreas, kidney and liver problems [Citation11,Citation16]. Furthermore, pentamidine treatment is complicated by the mode of drug administration which requires hospitalization of a patient for more than 7 days and therefore, creates significant logistical and societal problems in the remote rural areas of Africa, where HAT is endemic. This makes it difficult to implement with consistency because of poor health facilities and few healthcare personnel [Citation5,Citation12,Citation16,Citation17]. Pentamidine is not well absorbed through gastrointestinal tract (GIT) which leads to poor oral bioavailability and has a very slow rate of diffusion across the biological membranes and thus, making the concentrations not sufficient enough to affect the trypanosomes which advance to the central nervous system (CNS) [Citation11].
Figure 1. Pentamidine structure (Source [Citation11]).
![Figure 1. Pentamidine structure (Source [Citation11]).](/cms/asset/c8f967cf-7c62-4e7c-b5e6-089cf9fe380f/ianb_a_1596923_f0001_b.jpg)
Our work aimed at investigating the possibility of mitigating the toxicity of pentamidine and enhancing its transportation across the blood brain barrier (BBB) using nanoparticles. We loaded pentamidine in both polycaprolactone (PCL) nanoparticles and liposomes which are popular drug delivery vehicles for sustained and targeted drug delivery due to being relatively non-toxic, biodegradable, biocompatible and well tolerated by tissues [Citation18,Citation19]. The two loaded nanocarriers were characterized for physicochemical properties, examined for cytotoxicity and comparatively studied for the drug transportation efficiency across the endothelial monolayer tissue of the mouse BBB (b.End5 cells), an in vitro model for BBB.
Materials and methods
Materials for preparation of PCL nanoparticles
Polycaprolactone (PCL) polymer (2-oxepanone homopolymer, 6-caprolactone polymer; d = 1.146g/ml; average molecular weight =14,000; product of Japan in form of flakes) was a product from Sigma-Aldrich, St. Louis, MO, USA). The D-α-Tocopherol polyethylene glycol 1000 succinate (TPGS) (BioXtra, water soluble vitamin E conjugate; solubility: H2O 1g/10 ml; product of France) was a product from Sigma-Aldrich, St. Louis, MO, USA). Polysorbate (Tween® 80) (viscous liquid; P1754-25ML; CMC: 0.012 Mm (20–25OC) product of France) was a product from Sigma-Aldrich, MO, USA. Pentamidine isethionate salt (drug) (MW= 592.68 g/mol; P0547-250G; product of USA) was a product from Sigma-Aldrich, St. Louis, MO, USA). Chloroform was a product from Kimix Chemicals, Cape Town, SA). Materials for preparation of liposomes. L-a-phosphatidylcholine (Soy PC -95%); MW= 775.04; a product of USA from Avanti® polar lipids, Inc. 700 industrial Park Drive, Alabaster, Alabama. Cholesterol (Sigma Grade ≥93%; C8667-5G; MW = 386.65g/mol; was a product from Sigma-Aldrich, St. Louis, MO, USA. Phosphate Buffer Saline (PBS) (pH 7.4) was sourced from Nanotechnology innovation Centre (NIC), UWC laboratories and deionized water millipore (18.2 Ω) from Thermo Scientific) was used in all experiments.
Synthesis of pentamidine-loaded PCL nanoparticles
Pentamidine loaded PCL NPs were obtained by double solvent evaporation method as previously described by Ubrich and colleagues [Citation20] with some modifications. Briefly, a 1 mg/ml solution of pentamidine was made in a 0.1 mg/ml aqueous solution of TPGS prepared. Separately, a 5 mg/ml solution of PCL in chloroform was prepared. Thereafter, pentamidine-TPGS solution was poured to the PCL solution to mix the aqueous and oil phases. The container was placed on ice bath, and the primary emulsion (w/o) was formulated by homogenization using a high speed homogenizer (IKA® T18digital ULTRA TURRAX®; model: T18D) at 10 000 rpm for 3 min followed by probe tip sonication at 85% intensity for 3 min. To make the second emulsion, a 0.1 mg/ml aqueous solution of Tween® 80 was prepared and was added to the primary emulsion and homogenized at a speed of 3000 rpm for 3 min followed by probe sonication at 65% intensity for 30 s in order to form the double emulsion (w/o/w). To remove chloroform, the nanosuspension was evaporated under reduced pressure for 1 h at 40 °C. Nanoparticles were washed thrice with deionized water at 10,000 rpm, 10 min and 4 °C and collected after centrifugation.
Synthesis of pentamidine-loaded liposomes
Liposomes were formulated by thin lipid film hydration technique followed by extrusion as previously described by Begun and friends [Citation21] and Sha with colleagues [Citation22] with modifications. Briefly, L-phosphatidylcholine and cholesterol were weighed and mixed at the ratio of 4:1 and dissolved in chloroform. The formed emulsion was evaporated under vacuum (1 h, 55 °C) to remove chloroform and form a thin lipid film. Thereafter, 1 mg/ml solution of pentamidine isethionate were prepared in PBS at pH of 7.4. The mixture was then added to the flask containing the lipid film to hydrate the lipid film and encapsulate the drug. This was left for 30 min in a water-bath sonicator warmed at 25 °C. Thereafter, the contents were vortexed for 10 min to obtain the homogeneous solution. The suspension was extruded 10 times. Thereafter, the synthesized liposomes in the syringe were transferred to a beaker for purification. Liposomes were double purified with PBS (pH 7.4) by centrifugation (Eppendorf; centrifuge 5417R) at 10,000 rpm for 10 min at 4 °C.
Both pentamidine-loaded PCL nanoparticles and liposomes were cryoprotected with 10% sucrose, and freeze-dried for 24 h. Thereafter, size, polydispersity index (PDI) and zeta potential were determined using dynamic light scattering (DLS) techniques by a Zetasizer NanoZS90. Measurements were performed at 25 °C and 90° measurement scattering angle (10 runs). All measurements were recorded in triplicate as Mean diameter (nm) ± Standard Error (SE). Morphology was analyzed using Scanning Electron Microscope (LEO 1450 SEM, Zeiss).
Determination of pentamidine loading
The amount of loaded pentamidine was determined using HPLC coupled to a photo-diode array detector at ambient temperature. The Perkin Elmer (Shelton, CT, USA) HPLC-PDA system comprised of the Flexar LC Autosampler (100 ul injection loop), Flexar Solvent Manager 3-channel degasser, Flexar LC pump and the Flexar PDA Plus detector. Sample preparation was performed by breaking 1 mg of freeze dried pentamidine loaded nanoparticles in 1 ml of 0.1% formic acid (FA) warmed in a waterbath at 25 °C for 5 min. Thereafter, the solution was centrifuged at 10,000 rpm, 4 °C for 10 min and supernatant analyzed using HPLC. Working standards of concentration (0.01, 0.02, 0.1, 0.2, 0.5, 1 and 2) µg/ml were diluted from a 1 mg/ml pentamidine stock solution prepared in 0.1% FA to a final volume of 1,000 µl using 0.1% FA as diluent. 20 µl of sample or working standard was injected in triplicate at a flow rate of 1 ml/min onto an end-capped C8 reverse phase analytical LC column (Zorbax Eclipse XDB-C8 4.6 x 150 mm, 5 µm) with retention achieved by isocratic elution of the analyte at 10% of 60% aq. Acetonitrile and 90% of 0.1% FA. The absorbance maximum was determined at 265 nm and 270 nm. The standard curve of pentamidine was constructed at linearity over the range of 0.1 to 2 µg/ml (r2 = 0.9984) to quantify extracted pentamidine from PCL nanoparticles and liposomes. Loading was determined by using the following equation;
Determination of pentamidine release in vitro
To characterize drug release in vitro, 10 mg/ml concentrations of pentamidine-loaded freeze-dried PCL nanoparticles and freeze dried pentamidine-loaded liposomes were separately prepared in PBS (pH 7.4). Samples were incubated in a water bath at 37 °C and removed at time periods of 30 min, 1, 2, 6, 12, 24 and 48 h and centrifuged at 10,000 rpm, 4 °C for 10 min. Thereafter supernatants were frozen at –80 °C and later analyzed using the validated HPLC–PDA assay.
In vitro cell culture and cytotoxicity of PCL nanoparticles and liposomes
The b.End5 cells seeded at a density of 5 × 104 per well in supplemented Dulbecco’s Modified Eagles medium ham’s F12 (DMEM:F12) were cultured in a 6-well tissue culture plates and incubated at atmosphere of 37 °C and 5% CO2. Cells were incubated overnight to allow them to attach and expand to confluence. To determine the cytotoxic effect, in triplicate, three concentrations (1, 2.5 and 5 mg/ml) in supplemented DMEM:F12 of PCL nanoparticles, liposomes and free pentamidine were exposed to cells over a period of 96 h. After every 24 h interval, a Trypan blue exclusion assay (Sigma Cell Culture Reagents®, T-8154) was performed to evaluate % cell viability.
Pentamidine transport across b.End5 cell line
The immortalized mouse brain endothelioma (b.End5) cell line was purchased from the European Collection of Cell Cultures (ECACC, Sigma-Aldrich (Cat no. 96091930)) and was derived from brain endothelial cells of BALB/c mice. The b.End5 cells were seeded at the concentration of 5 × 105 cells per insert/well in a 24- well microtiter plate, on mixed cellulose esters Millicell® inserts with an area of 0.6 cm2 and then incubated at 37 °C and 5% CO2. Cells were allowed to attach to the bottom of the filter insert and allowed to grow to confluence in 24 h. Thereafter, they were removed from the incubator and allowed to acclimatize to room temperature for 20 min in a sterile laminar flow cabinet. Highest concentrations of nanoparticles and free drug which did not show effect on cells growth during cytotoxicity study of empty nanoparticles were used. Cells were exposed for 24 h to concentrations of 2.5 mg of PCL NPs loaded with 0.4 µg of pentamidine (0.4 µg/ml (w/v)), 1.1 mg of liposomes loaded with 0.2 µg of pentamidine (0.2 µg/ml (w/v)) and 0.4 µg of free pentamidine (0.4 µg/ml (w/v)). The permeability of the cell monolayer was assessed by measuring the Transendothelial Electrical Resistance (TEER) using a Millicell®- ERS voltohmmeter at 30 min, 1, 2, 3, 4, 6, 8, 12 and 24 h. All readings were taken in duplicate and the true resistance of endothelial monolayer was computed by subtracting the blank reading (insert without the cells) from the experimental sample reading. The reading was then standardized to square centimetres by multiplying the true resistance with the insert area (0.6 cm2). At the same time, 500 µl of sample were collected from the basolateral compartment at 2, 4, 6, 8, 12 and 24 h for quantification of pentamidine that crossed the monolayer from the apical side. Pentamidine from the samples was later analyzed with a validated HPLC-PDA and quantitation was carried out by extrapolating the peak areas against the concentrations to the calibration curve of pentamidine in the cell culture media with linearity over the range of 0.1 to 10 ppm (r2 = 0.9608). All experiments were conducted in triplicate.
Statistics
Statistical analysis was performed using One-way Analysis of Variance (1-way ANOVA) and paired t-test with GraphPad Prism 7.04 software. Statistical significant values were regarded when p values was <.05. Data were presented as Mean ± Standard Error (Mean ± SE).
Results
Characterization of PCL nanoparticles and liposomes
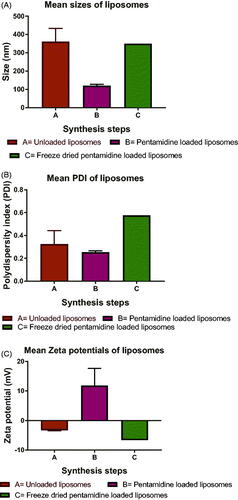
All synthesized particles were within nanoscale range where the smaller mean size was observed with pentamidine-loaded PCL NPs which was 267.58 ± 65.63 nm () and the larger size was observed with unloaded PCL NPs which was 344.18 ± 27.48 nm. The PDI was lower at all steps of synthesis with smaller mean PDI observed being 0.12 ± 0.05 () with unloaded PCL NPs and the higher was 0.3 ± 0.09 observed with freeze dried PCL NPs. The empty PCL NPs possessed a negative zeta potential with the lower mean zeta potential of –28.1 ± 4.19 mV () observed with loaded PCL NPs and the highest with freeze dried pentamidine-loaded PCL NPs which was –33.4 ± 8.12 mV. The mean PDI of 0.25 ± 0.15 of pentamidine loaded PCL NPs indicated uniformity of particle sizes produced and lower chance of aggregation of particles. The loaded PCL nanoparticles had a loading of pentamidine of 0.16 µg/mg (w/w). Likewise, liposomes had sizes within the nanoscale range with smaller mean size observed with pentamidine-loaded liposomes which was 119.61 ± 14.31 nm () and the larger mean size was observed with unloaded liposomes which was 360.84 ± 125.93 nm. The mean size of pentamidine loaded liposomes was 119.61 ± 14.31 nm and Agrawal [Citation23] reported that liposomes with the size range below 200 nm were easily able to cross the BBB. The mean PDI of 0.25 ± 0.02 () observed with loaded liposomes indicated monodispersion of liposomes in suspension, unlikely to aggregate and stable. Both positive and negative zeta potential were observed. The mean surface charge of these liposomes was positive 11.78 ± 10.15 mV () which suggested that some pentamidine was adsorbed on the surface of liposomes. Post freeze drying we observed the zeta potential changing to –6.6 mV which might inform that because pentamidine is water-soluble, the traces might have come into contact with bound water and dissolved in water during freezing process and then was taken out with water during secondary drying in process of freeze drying [Citation24]. Loaded liposomes had a loading of 0.178 µg/mg (w/w). Both PCL nanoparticles and liposomes presented good physicochemical properties of nanomaterials and we considered them suitable carriers of drug across the BBB. There was no statistical significant difference between the size means of PCL NPs and liposomes (paired t-test, p = .5928). This presents the double solvent evaporation and thin film hydration techniques being suitable for synthesis of drug carriers for BBB transportation.
Figure 2. Particle physicochemical properties of unloaded, pentamidine-loaded and freeze dried pentamidine-loaded PCL nanoparticles. (A) Sizes, (B) PDIs, (C) Zeta potentials. Data is presented as mean ± SE. Sample size (n = 6). No statistical differences were found at p < .05.
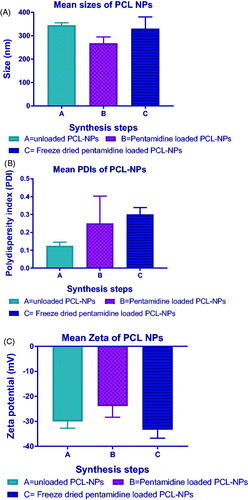
Figure 3. Liposomal particle physicochemical properties of unloaded, pentamidine-loaded and freeze dried pentamidine-loaded liposomes. (A) Sizes, (B) PDI, (C) Zeta potential. Data is presented as mean ± SE. Sample size (unloaded and loaded liposomes n = 3 and freeze-dried n = 1(pooled sample)). No statistical differences were found at p < .05.
SEM revealed smooth spherical shapes for both PCL nanocapsules and liposomes (). Spherical shaped or spheres are ease to synthesis and have been studied for years. They are quickly up taken by microphages and stay for shorter time in circulation as compared to non-spherical nanoparticles [Citation25,Citation26]. However, the sizes observed also correlated well with the hydrodynamic diameters measured by the Zetasizer ().
Figure 4. These are electron micrographs of freeze dried PCL nanoparticles (A) and freeze dried liposomes (B) generated by SEM. The legends were generated by SEM and all bars calibrated at 1 µm.

The amounts of pentamidine released were extrapolated from the calibration curve of pentamidine in PBS. Results show that over a period of 24 h, 12.13 ± 0.21% and 22.21 ± 0.53% of pentamidine was released from PCL nanoparticles and liposomes respectively. Statistical difference between the mean percentages of PCL NPs and liposomes pentamidine release was significant (paired t-test, p = .0002).
Toxicological effect
It was important to screen for the appropriate concentration of both unloaded and loaded nanoparticles with no toxic effect to b.End5 cells to use for transportation studies in vitro. The survival rates were determined by using Trypan Blue exclusion assay. The study indicate that unloaded PCL nanoparticles exposed at highest concentration of 5 mg/ml () (p = .2072) and unloaded liposomes at concentrations of 5 mg/ml and 2.5 mg/ml () (p = .0378) had cytotoxic effect and consequently inhibited the growth of the b.End5 cells over a period of 96 h. In these concentrations hardly growing cells were observed at 48 h to 96 h. However, statistically, the difference in cell viability following exposure of three concentrations of unloaded PCL NPs was insignificant (p = .2072). Conversely, pentamidine-loaded PCL nanoparticles exposed at the concentration of 2.5 mg/ml (loading 0.4 µg pentamidine) () (p = .9916), pentamidine-loaded liposomes exposed at higher concentrations of 1 mg/ml (loading 0.2 µg pentamidine) () (p = .9023) and free pentamidine of up to 0.4 µg () did not affect cells viability. However, free pentamidine exposed at higher level concentration of 200 µg/ml and 400 µg/ml had severe cytotoxic effect on cells (p = .0006) such that hardly any viable cells were observed after 24 h exposure (). However, we anticipated that loaded nanoparticles in concentrations above the screened ones would result into cytotoxic effect as observed with unloaded particles and would not be safe to use such concentrations for transportation study.
Figure 5. Shows the toxic effect of 5 mg/ml concentration of unloaded PCL NPs on the growth of b.End5 cells. Data is presented as mean ± SE, n = 3. No statistical differences were found at p < .05.
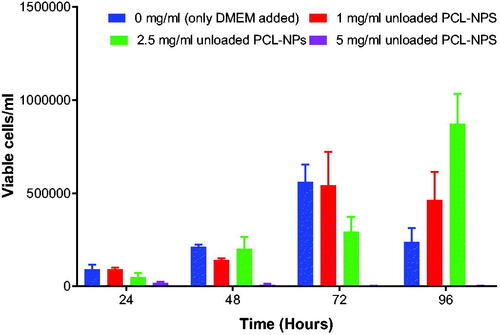
Figure 6. Shows the effect of different concentrations of pentamidine-loaded NPs on the growth b.End5 cells. Data is presented as mean ± SE, n = 3. No statistical differences were found at p < .05.
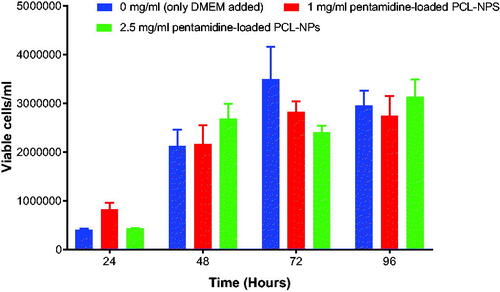
Figure 7. Shows toxic effect of 2.5 mg/ml and 5 mg/ml concentrations of pentamidine-loaded liposomes on the growth of b.End5 cells. Data is presented as mean ± SE, n = 3. Statistical significant difference was found with 2.5 mg/ml concentration at p < .05.
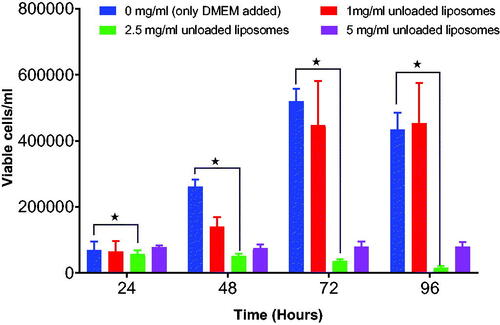
Figure 8. Shows the effect of different concentrations of pentamidine- loaded liposomes on growth of b.End5 cells. Data is presented as mean ± SE, n = 3. No statistical difference was found at p < .05.
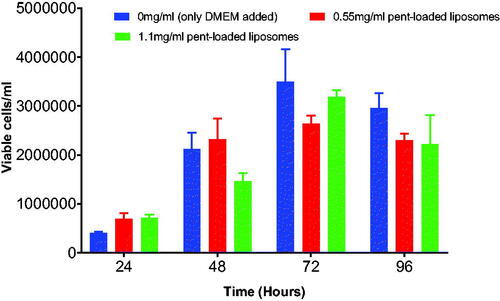
Figure 9. Shows the effect of different concentrations of free pentamidine on the growth of b.End5 cells. 200 µg/ml and 400 µg/ml were severely toxic to cells that no cells could grow. Both concentrations had statistical significant difference at p < .05. Data is presented as mean ± SE of mean, n = 3.
Transportation of pentamidine across the in b.End5 cells
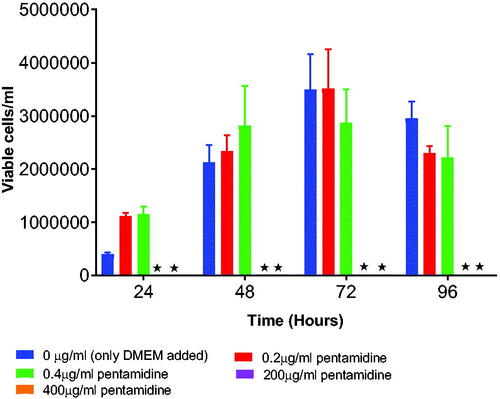
TEER was measured to assess the integrity of the cell monolayer following drug exposure to either pentamidine, or pentamidine loaded PCL nanoparticles or liposomes. When pentamidine-loaded PCL NPs, pentamidine-loaded liposomes, free pentamidine and media as control were added in wells, TEER values dropped in the first two hours which signify loosening of the tight junctions but then values raised with time which imply increased resistance of the monolayer restricting penetration of molecules through the tight junctions over 24 h. The TEER values raised and from the eighth hour of exposure resistance was constantly sustained. TEER values for the control were observed to raise shortly and continually decreased with time (). However, the TEER values differences observed between the control and liposomes (p = .9458), control and PCL NPs (p = .2972), control and free pentamidine (p = .3014), Liposomes and PCL NPs (p = .6076), liposomes and free pentamidine (p = .6131), PCL NPs and free pentamidine (p = .9999) were all statistically insignificant indicating that the cells monolayer was intact and sensitively restricting the penetration of any molecule across it.
Figure 10. The effects of pentamidine-loaded PCL nanoparticles, pentamidine loaded liposomes and free pentamidine on the TEER of b.End5 cell monolayers. Data is presented as mean ± SE, n = 3. No statistical difference was found at p < .05.
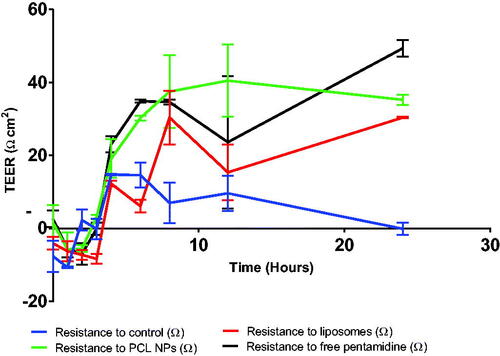
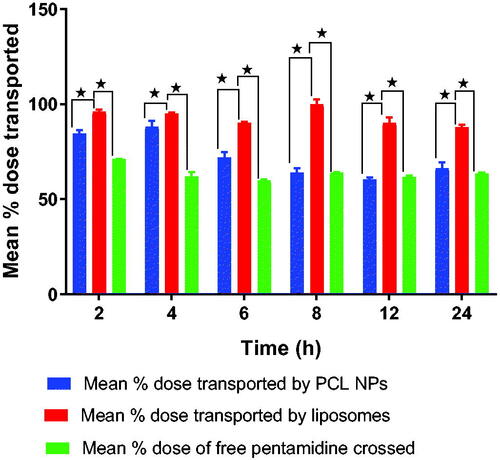
The quantities of pentamidine collected from the basolateral compartment were analyzed by HPLC–PDA. Comparatively, liposomes were observed to deliver more dose of pentamidine to the basolateral compartment compared to PCL NPs and movement of free pentamidine () which correlates well with what was observed with TEER responses as well as drug release profile of liposomes and PCL NPs. After 24 h, liposomes could transport 87 ± 0.01% of the pentamidine dose, while PCL NPs could transport 66 ± 0.1% of the dose and free pentamidine solution could deliver 63 ± 0.25% of the dose. The percent dose transport difference of liposomes against PCL NPs was statistically significant (p = .0006) and against free drug (p = .0001) but there was no statistical significant difference of percent dose transport between PCL NPs and movement of free drug (p = .1281). Overtime, the delivered quantities were observed to decrease which is also a reflection of the TEER values profile (see ) which first dropped signifying allowed transportation of molecules through the BBB and then slowly raised indicating strong restriction movement of molecules across the barrier. We may explain that free drug, liposomes and nanoparticles were trapped within the monolayer cells as time went on but also we think transportation of liposomes and nanoparticles utilized mainly the transport mediated mechanism such as endocytosis or transcytosis. Dose delivered differences were statistically significant (p = .0001).
Figure 11. Shows the percentage dose of free pentamidine, pentamidine-loaded PCL NPS and pentamidine-loaded liposomes transported across the cells monolayer. Here, we were testing the transportation ability of liposomes and PCL NPs nanocarriers to ferry pentamidine across the BBB as compared to free movement of pentamidine. Data is presented as mean ± SE, n = 3. Transport means differences between liposome and PCL NPs, and liposome and free drug were found statistically significant at p < .05.
Discussion
Several techniques have been developed and became successful in loading drugs into the polymeric nanoparticles for drug delivery, and the choice for the method of encapsulation usually depends on the physicochemical properties of the drug itself [Citation27]. Using the double emulsion solvent evaporation as a technique to encapsulate pentamidine, a water-soluble drug in this work was due to the fact that the double emulsion techniques was previously shown to improve the drug encapsulation and minimize the escape of the hydrophilic drug from the aqueous core which was our aim [Citation20,Citation27]. Furthermore, two important surfactants (Polysorbate 80 and TPGS) were used. TPGS has been shown to increase the loading ability of hydrophilic drugs into the polymeric nanoparticles and polysorbate 80 has been reported to help synthesis of small sized nanoparticles and also increase the ability of nanoparticles to cross the BBB [Citation28–31]. Hans and Lowman [Citation28] stated that in blood circulation, nanoparticles coated with polysorbate 80 adsorbs apolipoprotein E on the surface of the coating, the apolipoprotein E mimics the low density lipoprotein (LDL) causing the particles to be transported via the LDL receptors into the brain. Our production of nanoparticles involved two rounds of homogenization and sonication. We successfully produced spherical polycaprolactone nanoparticles with suitable characteristics. The mean size of the dried loaded particles was 329.62 ± 121.91 nm which was desirable and hypothesized to be able to improve drug penetration across the BBB. The particles were also observed to have higher negative charge of –33.4 ± 8.12 which could induce the repulsive force to the particles when they get into contact with the biological membranes which are negatively charged. However, we suggested that nanoparticles being coated with polysorbate 80 which adsorbs apolipoprotein E would increase the penetration of nanoparticles across the BBB [Citation29,Citation32].
Drug release showed that pentamidine released was slow and constant over 24 h. Drug diffusion and degradation of PCL polymer is usually slow and the release would be expected to start slowly and extend as the polymer erodes [Citation33,Citation34]. However, the constant release showed that PCL hydrolyzes slowly and thus sustain slow drug release over a longer period as observed [Citation33]. In addition, the longer release might be caused by poor drug loading and thus slower release rate [Citation28] we observe. This could be good for the release of toxic drugs like pentamidine but also we think that optimization of the drug loading protocols could help to improve the encapsulation efficiency.
Liposomes are made of phospholipids and hence considered biocompatible, biodegradable and less toxic [Citation21]. Because liposomes are amphiphilic in nature they are appropriate for encapsulation of drugs and delivery of both hydrophilic and hydrophobic drugs [Citation35]. Therefore, liposomes were also an appropriate choice for encapsulation of pentamidine in this study. The aim was to ascertain which of the nanocarriers between polymeric nanoparticles and liposomes would be efficient in transporting pentamidine across in vitro endothelial cells of the BBB. We successfully formulated liposomes of the desirable characteristics for delivery of pentamidine. We hypothesized that with the increased concentration of liposomes on the luminal side of the endothelial membrane, liposomes would be easily transported across the barrier.
This study demonstrated that higher concentrations of both PCL nanoparticles and liposomes may inhibit cellular growth. We observed that PCL NPS at a dose of 5 mg/ml and liposomes at 2.5 mg/ml and 5 mg/ml were able to inhibit multiplication of cells and caused death to cells ( and ). However, this study alone cannot justify the real cause of cytotoxicity. Nevertheless, polysorbate 80 and TPGS may be toxic if used at high concentrations.
TEER values are a good indicator when the integrity and permeability of the biological barrier like the BBB is assessed [Citation36]. When TEER values rise, this typically indicates that the barrier restricts the movement of any solute through the tight junction. On the other hand, a drop in TEER values is also a good indicator that the barrier allows the passive movement of molecules through the tight junction. The average size of pentamidine loaded liposomes was about 119 nm and we suggest that most of the liposomes together with other factors penetrated the barrier through the paracellular pathway. However, liposomes are lipophilic in nature and that might have added the advantage for them to cross the barrier. Restriction to PCL nanoparticles and free drug as assessed by TEER values was comparatively the same. In our experiment, we observed that liposome nanocarriers were better and able to transport more dose percentage of pentamidine compared to PCL NPs and the movement of free drug. Nevertheless, the two nanocarriers are promising delivery systems which can improve the delivery of pentamidine drug across the BBB. Therefore, we recommend future studies to evaluate these nanocarriers in vivo.
Disclosure of interest
The authors report no conflict of interest.
Acknowledgements
The authors would like to thank the UWC, South Africa and particularly the Schools of Pharmacy and Life Sciences for provision of facilities and laboratory space for carrying out this research. Furthermore, authors extend thanks to Dr. Cummings and Mr. Earl from the School of Physics for helping the morphological characterization of nanoparticles and liposomes by SEM. We also thank NMAIST for providing permission to undertake this study at the UWC. GO acknowledges the Tanzanian Higher Education Students’ Loan Board (HESLB) and NMAIST CREATES Program for providing financial support that enabled undertaking of this project.
Related Research Data
References
- Denise H, Barrett MP. Uptake and mode of action of drugs used against sleeping sickness. Biochem Pharmacol. 2001;61:1–5.
- Thuita JK, Wolf KK, Murilla GA, et al. Safety, pharmacokinetic, and efficacy studies of oral DB868 in a first stage vervet monkey model of Human African Trypanosomiasis. PLoS Negl Trop Dis. 2013;7:e2230.
- Aksoy S, Buscher P, Lehane M, et al. Human African trypanosomiasis control: achievements and challenges. PLoS Negl Trop Dis. 2017;11:1–6.
- Aksoy S, Attardo G, Berriman M, et al. Human African trypanosomiasis research gets a boost: unraveling the Tsetse genome. PLoS Negl Trop Dis. 2014;8:6–8.
- Babokhov P, Sanyaolu AO, Oyibo WA, et al. A current analysis of chemotherapy strategies for the treatment of human African trypanosomiasis. Pathog Glob Health. 2013;107:242–252.
- Mäser P, Lüscher A, Kaminsky R. Drug transport and drug resistance in African trypanosomes. Drug Resistance. Drug Resist Updat. 2003;6:281–290.
- Sanderson L, Dogruel M, Rodgers J, et al. Pentamidine movement across the murine blood-brain and blood-cerebrospinal fluid barriers: effect of trypanosome infection, combination therapy, P-glycoprotein, and multidrug resistance-associated protein. J Pharmacol Exp Therapeut. 2009;329:967–977.
- Etchegorry MG, Helenport JP, Pecoul B, et al. Availability and affordability of treatment for Human African Trypanosomiasis. Trop Med Int Health. 2001;6:957–959.
- Arias JL, Unciti-Broceta JD, Maceira J, et al. Nanobody conjugated PLGA nanoparticles for active targeting of African Trypanosomiasis. J Control Release. 2015;197:190–198.
- Aksoy S, Maudlin I, Dale C, et al. Prospects for control of African trypanosomiasis by tsetse vector manipulation. Parasitol Today. 2001;17:29–35.
- Porcheddu A, Giacomelli G, De Luca L. New pentamidine analogues in medicinal chemistry. Curr Med Chem. 2012;19:5819–5836.
- Baker N, de Koning HP, Mäser P, et al. Drug resistance in African trypanosomiasis: the melarsoprol and pentamidine story. Trends Parasitol. 2013;29:110–118.
- Lin JM-H, Shi RJ, Lin ET. High performance liquid chromatographic determination of pentamidine in plasma. J Liq Chromatogr. 1986;9:9:2035–2046.
- Soeiro MNC, Werbovetz K, Boykin DW, et al. Novel amidines and analogues as promising agents against intracellular parasites: a systematic review. Parasitology. 2013;140:929–951.
- Burri C. Chemotherapy against human African trypanosomiasis: is there a road to success?. Parasitology. 2010;137:1987–1994.
- Wilson WD, Tanious FA, Mathis A, et al. Antiparasitic compounds that target DNA. Biochimie. 2008;90:999–1014.
- Wenzler T, Boykin DW, Ismail MA, et al. New treatment option for second-stage African sleeping sickness: in vitro and in vivo efficacy of aza analogs of DB289. Antimicrob Agents Chemother. 2009;53:4185–4192.
- Paul M, Laatiris A, Fessi H, et al. Pentamidine-loaded poly (D, L-lactide) nanoparticles: adsorption and drug release. Drug Dev Res. 1998;43:98–104.
- Dua JS, Rana AC, Bhandari A. K. Review article liposome: methods of preparation and applications. Int J Pharmaceut Stud Res. 2012;III:7.
- Ubrich N, Bouillot P, Pellerin C, et al. Preparation and characterization of propranolol hydrochloride nanoparticles: a comparative study. J Control Release. 2004;97:291–300.
- Begum MY, Sudhakar M, Abbulu K. Flurbiprofen-loaded stealth liposomes: studies on the development, characterization, pharmacokinetics, and biodistribution. J Young Pharm. 2012;4:209–219.
- Sha X, Guo J, Chen Y, et al. Effect of phospholipid composition on pharmacokinetics and biodistribution of epirubicin liposomes. J Liposome Res. 2012;22:80–88.
- Agrawal M, Ajazuddin, Tripathi DK, et al. Recent advancements in liposomes targeting strategies to cross blood-brain barrier (BBB) for the treatment of Alzheimer’s disease. J Control Release. 2017;260:61–77.
- Abdelwahed W, Degobert G, Stainmesse S, et al. Freeze-drying of nanoparticles: formulation, process and storage considerations. Adv Drug Deliv Rev. 2006;58:1688–1713.
- Ta HT, Truong NP, Whittaker AK, et al. The effects of particle size, shape, density and flow characteristics on particle margination to vascular walls in cardiovascular diseases. Expert Opin Drug Deliv. 2018;15:33–45.
- Truong NP, Whittaker MR, Mak CW, et al. The importance of nanoparticle shape in cancer drug delivery. Expert Opin Drug Deliv. 2015;12:129–142.
- Cohen-Sela E, Chorny M, Koroukhov N, et al. A new double emulsion solvent diffusion technique for encapsulating hydrophilic molecules in PLGA nanoparticles. J Control Release. 2009;133:90–95.
- Hans ML, Lowman AM. Biodegradable nanoparticles for drug delivery and targeting. Curr Opin Solid State Mater Sci. 2002;6:319–327.
- Göppert TM, Müller RH. (Polysorbate-stabilized solid lipid nanoparticles as colloidal carriers for intravenous targeting of drugs to the brain: comparison of plasma protein adsorption patterns. J Drug Target. 2005;13:179–187.
- Limayem Blouza I, Charcosset C, Sfar S, et al. Preparation and characterization of spironolactone-loaded nanocapsules for paediatric use. Int J Pharmaceut. 2006;325:124–131.
- Guo Y, Luo J, Tan S, et al. The applications of vitamin E TPGS in drug delivery. Eur J Pharm Sci. 2013;49:175–186.
- Chigumira W, Maposa P, Gadaga LL, et al. Preparation and evaluation of pralidoxime-loaded PLGA nanoparticles as potential carriers of the drug across the blood brain barrier. J Nanomater. 2015;2015:1.
- Bordes C, Fréville V, Ruffin E, et al. Determination of poly (Ɛ-caprolactone) solubility parameters: application to solvent substitution in a microencapsulation process. Int J Pharmaceut. 2010;383:236–243.
- Iqbal M, Valour JP, Fessi H, et al. Preparation of biodegradable PCL particles via double emulsion evaporation method using ultrasound technique. Colloid Polym Sci. 2015;293:861–873.
- Lopes LB, Scarpa MV, Silva GVJ, et al. Studies on the encapsulation of diclofenac in small unilamellar liposomes of soya phosphatidylcholine. Colloids Surf B Biointerfaces. 2004;39:151–158.
- Srinivasan B, Kolli AR, Esch MB, et al. TEER measurement techniques for in vitro barrier model systems. J Lab Autom. 2015;20:107–126.