
Abstract
Choriocarcinoma is a rare gestational trophoblastic tumour. Propofol is one of the commonly used intravenous anaesthetic agents and exerts an anti-tumour role in multiple cancers. However, the effect of propofol on choriocarcinoma has not been well explored. We investigated the effect of propofol on JEG-3 cells. The effects of propofol on cell proliferation and metastasis were determined. The expression of miR-495 was suppressed while Twist1 overexpressed. Cell viability, migration and invasion were respectively measured using CCK-8 assay and transwell migration/invasion assay. The expression of miR-495, AKT1 and Twist1 was detected using qRT-PCR. Western blot analysis was performed to assess the expression levels of EMT- and proliferation-related factors, and TGF-β pathway proteins. Propofol treatment significantly inhibited cell proliferation, migration and invasion, and reduced the releasing of EMT. miR-495 was up-regulated by profocol, and miR-495 inhibition reversed the effect of propofol on cell proliferation and metastasis in JEG-3 cells. Furthermore, propofol decreased the expression of AKT1 and Twist1 through up-regulating miR-495, and Twist1 participated in the regulation of propofol on TGF-β signalling inactivation in JEG-3 cells. This study demonstrates that propofol suppresses cell proliferation and metastasis through up-regulation of miR-495 and down-regulation of Twist1 in JEG3 cells.
Propofol inhibits proliferation and metastasis in JEG-3 cells;
miR-495 is up-regulated by propofol in JEG-3 cells;
miR-495 suppression blocks the effect of propofol in JEG-3 cells;
Propofol decreases Twist1 expression through up-regulating miR-495;
Propofol inhibits TGF-β signalling via regulation of Twist1.
Highlights
Introduction
Gestational trophoblastic disease (GTD) is a unique gynaecological disease derived from the placenta and comprises of gestational trophoblastic neoplasm (GTN), hydatidiform mole (HM), nonneoplastic lesions and abnormal villous lesions [Citation1]. Choriocarcinoma, one of the most aggressive gestational trophoblastic neoplasm, has drawn considerable attention since the first report of a metastatic choriocarcinoma patient being cured by methotrexate in 1956 [Citation2]. Although choriocarcinoma is not a common disease, it can spread rapidly and has a mortality rate of nearly 100% if not been treated in time [Citation3]. Recently, great progress has been made in understanding the physiological and pathological processes of human trophoblasts, yet the pathogenesis of choriocarcinoma is still not completely understood due to the increasing rarity of the disease and the lack of an animal model [Citation4,Citation5].
microRNAs (miRNAs), a new class of endogenous non-coding RNAs, are involved in post-transcriptional gene regulation by binding to a target site in the 3’-UTR of target mRNAs and result in mRNA degradation or inhibition of translation [Citation6]. It has been reported that miRNAs functioned as either oncogenes or tumour suppressors through specifically regulating the expression of their target genes depending on cellular context [Citation7]. Previous studies have revealed that miR-495 was involved in multiple cancers, including breast cancer, ovarian cancer, endometrial cancer, gastric cancer, hepatocellular cancer, oesophagal squamous cell carcinoma and gallbladder cancer [Citation8–12]. For example, miR-495 expression was down-regulated in oesophagal squamous cell carcinoma (ESCC) tumour specimens and its overexpression inhibited ESCC cell proliferation, migration and invasion through directly targeting of AKT1 [Citation8]. In breast cancer stem cells, miR-495 was up-regulated by E12/E47 and enhanced oncogenesis and hypoxia resistance via the down-regulation of E-cadherin and REDD1 [Citation10]. Meanwhile, miR-495 acts as a tumour suppressor to inhibit gastric cancer cell migration and invasion by regulating phosphatase of regenerating liver (PRL)-3 [Citation13]. However, the specific function of miR-495 in the progression of choriocarcinoma still remains unclear.
Propofol, 2,6-diisopropylphenol, is extensively commonly used for induction and maintenance of anaesthesia [Citation14]. Recently, increasing evidence reveal that propofol has the ability to modulate the motility, proliferation, and invasion of cancer cells in vitro and in vivo [Citation15–17]. Therefore, propofol might be an effective drug for cancer treatment [Citation18,Citation19]. However, there is no sufficient information on the effect of propofol in choriocarcinoma.
The present study aimed to explore the effect of propofol in cell proliferation and metastasis in choriocarcinoma cell line, JEG-3 cells and the further underlying mechanisms. We found that propofol inhibited JEG-3 cell proliferation, migration and invasion, and epithelial-mesenchymal transition (EMT) process, and the underlying mechanism might be through up-regulation of miR-495.
Materials and methods
Cell culture and treatment
The human choriocarcinoma-derived placental JEG-3 cell line was purchased from the American Type Culture Collection (ATCC; Rockville, MD, USA) and cultured in Dulbecco’s modified Eagle Medium (DMEM; Sigma-Aldrich, St. Louis, MO, USA) without phenol red and supplemented with 10% charcoal-stripped fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA) (and thus depleted of steroid hormones), 100 UI/ml penicillin and 100 g/ml streptomycin (Gibco). The JEG-3 cells were cultured in a humidified environment with 5% CO2 and 95% air at 37 °C. The JEG-3 cells were incubated with 10 ng/ml of transforming growth factor-β (TGF-β; PeproTech, Rocky Hill, USA) for 24 h, which was used for inducing EMT [Citation20].
Transfection and stable cell line establishment
miR-495 inhibitor and the corresponding negative control (NC) were synthesized using GenePharma (Shanghai, China). JEG-3 cells were seeded on 6-well plates and were transfected with miR-495 inhibitor and NC vectors when it reached 50% influence. Cell transfections were conducted using Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol.
For the analysis of Twist1 functions in JEG-3 cells, the whole sequence of Twist1 transcript was cloned into pEX vector and referred as pEX-Twist1 (GenePharma). The Lipofectamine 3000 reagent (Invitrogen) was used for the cell transfection according to the manufacturer’s instructions. A non-targeting sequence of plasmid was used as NC of pEX-Twist1 and referred as pEX. The stably transfected cells were selected by the culture medium containing 0.5 mg/ml G418 (Sigma-Aldrich). After approximately 4 weeks, G418-resistant cell clones were established.
CCK-8 assay
JEG-3 cells were seeded in 96-well plates with 5 × 103 cells/well, and cell viability was measured using a Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, Gaithersburg, MD). Briefly, after treatment with different concentrations of propofol (10 nM, 100 nM, 1 μM and 10 μM), 10 μl CCK-8 solution was added to the culture medium, and the cells were incubated for another 1 h at 37 °C in humidified 95% air and 5% CO2. The absorbance was measured at 450 nm using a Microplate Reader (Bio-Rad Laboratories, Orlando, FL, USA).
For determining role of miR-495 in JEG-3 cell viability, JEG-3 cells were transfected with miR-495 inhibitor and the corresponding NC. Then, transfected cells or non-transfected cells were treated with 1 μM propofol for 24 h, and cell viability was evaluated as mentioned earlier.
Ki67+ cell proliferation
The specimens were fixed with 10% neutral formalin, embedded with paraffin, and sliced continuously. Immunohistochemistry was used to detect the expression of Ki67. PBS was used as negative control group instead of primary antibody. Paraffin sections were dewaxed and gradient hydrated. After high-pressure original restoration, the paraffin was incubated with 3% hydrogen peroxide for 20 min in a warm and cold room. Later, normal sheep serum was added and incubated for another 20 min. Then, add 1:100 working concentration of Ki67 antibody; placed the concentration overnight in a 4 °C temperature environment, and add 1:100 ratio of biotin labelling at a room temperature for 15 min incubation; then, add 1:100 ratio of avidin horseradish peroxidase tags after incubation, and again after 15 min use neutral resin sealing piece for dyeing process.
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated and extracted from cells using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. For the level of miR-495 detection, reverse-transcribed complementary DNA was synthesized with the PrimeScript RT reagent Kit (TaKaRa, Dalian, China), and qRT-PCR was performed with SYBR Premix ExTaq (TaKaRa) with the Stratagene Mx3000P real-time PCR system (Agilent Technologies, Inc., Santa Clara, CA, USA). Expression levels were normalized against the endogenous snRNA U6 control. For mRNA analyses, cDNA was synthesized using Moloney murine leukaemia virus reverse transcriptase (Promega, Madison, WI, USA). qRT-PCR was performed with SYBR Premix ExTaq with the Stratagene Mx3000P real-time PCR system (Agilent Technologies). GAPDH was used as internal controls for mRNA quantification. The relative expression ratio was calculated using the 2−ΔΔCT method. The primer of AKT1 is forward: 5'-ACT CTT TCC AGA CCC ACG AC-3', reverse: 5'-CCA GGG CTG ACA CAA TCT CA-3'; Twist1, forward: 5'-CGC CCC GCT CTT CTC CTCT-3′, Reverse: 5′-GAC TGT CCA TTT TCT CCT TCT CTG-3’; GAPDH, forward: 5'-TCA ACG ACC ACT TTG TCA AGC TCA-3', Reverse: 5'-GCT GGT GGT CCA GGG GTC TTA CT-3'; miR-495, forward: 5’-ACA CTC CAG CTG GGG AAG TTG CCC ATG TTA-3’ reverse: 5'-TGG TGT CGT GGA GTCG-3′; U6, Forward: 5’-CTC GCT TCG GCA GCA CA-3’, reverse: 5’-AAC GCT TCA CGA ATT TGC GT-3’; miR-495 inhibitor, 5’-AAG AAG UGC ACC AUG UUU GUUU-3’.
Migration and invasion assay
Cell migration was determined using a modified two-chamber migration assay with a pore size of 8 μm (Corning, Lowell, MA, USA). For migration assay, cells suspended in 200 μl of serum-free medium were seeded on the upper compartment of 24-well Transwell culture chambers while the lower compartment was filled with 600 μl of complete medium. After incubation at 37 °C for 48 h, cells were fixed with methanol. Non-traversed cells were removed from the upper surface of the filter carefully with a cotton swab. Traversed cells on the lower side of the filter were stained with 0.1% crystal violet (Sigma-Aldrich) and counted by an inverted microscope (Nikon Corp., Tokyo, Japan). Cell invasion was conducted in the similar process as cell migration, but the inserts were coated with 20 μg of Matrigel (BD Biosciences, Bedford, MA, USA).
Western blot
The protein used for western blot was extracted using RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) supplemented with protease inhibitors (Roche, Basel, Switzerland). Quantification of proteins was assessed using BCA™ Protein Assay Kit (Pierce, Appleton, WI, USA). Equivalent proteins were loaded and separated using a Bio-Rad Bis-Tris Gel system according to the manufacturer’s instructions and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA). Subsequently, the membranes were blocked with 5% BSA (Roche) in Tris-buffered saline containing 1% Tween-20 (TBST). Primary antibodies: anti-GAPDH (#2118), anti-E-cadherin (#3195), anti-N-cadherin (#13116), anti-Vimentin (#5741), anti-PCNA (#13110), anti-Cyclin A (#4656), anti-Cyclin E1 (#20808), anti-Cyclin D1 (#2978), anti-CDK4 (12790), anti-Twist1 (#46702), anti-Smad2/3 (#8685), anti-p-Smad2/3 (#8828; Cell Signaling Technology, Beverly, MA, USA) and anti-CDK1 (ab18; Abcam, Cambridge, United Kingdom) were prepared in 5% BSA at dilutions according to the manufactures’ instructions. The blots were incubated with primary antibodies overnight at 4 °C. Afterwards, the blots were rinsed with TBST for three times and further incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG or anti-mouse IgG (Sigma-Aldrich) at a 1:5000 dilution for 2 h at room temperature. After being washed with TBST, the blots were developed with ECL solution (Pierce) and visualized using Image Lab™ Software (Bio-Rad, Hercules, CA, USA).
Statistical analysis
All experiments were repeated three times. The results were presented as the mean ± standard deviation (SD). Statistical analyses were performed using Graphpad 6.0 statistical software (GraphPad Software, San Diego, CA, USA). Statistical analysis was carried out using the Student’s t-test for comparisons of two groups, and data with three groups or more were analyzed using a one-way analysis of variance (ANOVA) followed by Tukey post-hoc test. A p value of <.05 was considered to indicate a statistically significant result.
Results
Propofol suppresses cell proliferation and metastasis in JEG-3 cells
First, JEG-3 cells were treated with different concentrations of propofol (10 nM, 100 nM, 1 μM and 10 μM), and the cell viability was assessed. As shown in , propofol significantly decreased cell viability in a dose-dependent manner in JEG-3 cells (p < .05, p < .01, or p < .001). Since 1 μM resulted in a significant decrease of cell viability in JEG-3 cells, 1 μM was selected as an effective concentration for the following experiments. Then, we detected the effect of propofol on cell migration and invasion. 1 μM propofol significantly declined the numbers of migrated and invasive cells () (p < .05, or p < .01). Members of the transforming growth factor-β (TGF-β) superfamily have been implicated as major induction signals of EMT [Citation21]. We further explored the regulatory functions of propofol on EMT process in JEG-3 cells by analyzing the protein expression levels of EMT-related proteins (E-cadherin, N-cadherin and Vimentin). Western blot analysis revealed that the expression of epithelial cell biomarker E-cadherin was down-regulated, and the levels of interstitial cell biomarker N-cadherin and Vimentin were up-regulated after TGF-β1 exposure, which indicated that EMT was induced by TGF-β1. Propofol inhibited the EMT process, as evidenced by increasing of E-cadherin and decreasing of N-cadherin and Vimentin levels (). Furthermore, propofol reduced the protein expression of proliferation-related factors, such as PCNA, Cyclin A, Cyclin E1, Cyclin D1, CDK1 and CDK4 (). These results indicated that propofol inhibits cell proliferation, migration and invasion, and EMT process in JEG-3 cells.
Figure 1. Propofol suppresses cell proliferation and metastasis in JEG-3 cells. JEG-3 cells were treated with various concentration of propofol (10 nM, 100 nM, 1 μM and 10 μM). (A) Cell viability was assessed by CCK-8 assay. (B) Relative migration and (C) invasion was measured using Transwell analysis. (D) The expression of EMT-related factors in JEG-3 cells was detected using western blot. (E) Western blot analysis of cell proliferation-regulated factors in JEG-3 cells. *p < .05, **p < .01, ***p < .001.
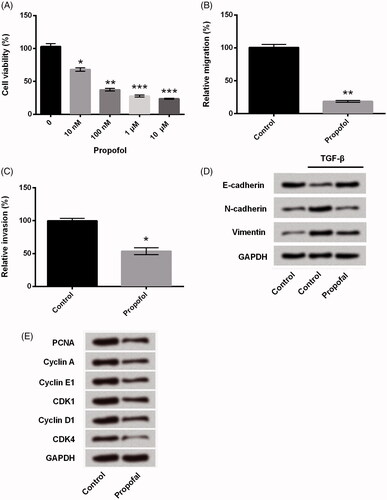
Propofol up-regulates miR-495 expression in JEG-3 cells
Considering that miR-495 plays a critical role in the development of oncogenesis via regulation of cell growth and metastasis [Citation8,Citation11,Citation12]. We explored whether the regulatory role of propofol was connected with miR-495. As shown in , the expression of miR-495 was significantly up-regulated in JEG-3 cells by treatment with propofol (p < .01), which suggested that miR-495 might be involved in the regulation of propofol in JEG-3 cells.
Figure 2. Propofol up-regulates miR-495 expression in JEG-3 cells. After treated with propofol (1 μM), the expression of miR-495 in JEG-3 cells was detected using qRT-PCR. **p < .01.
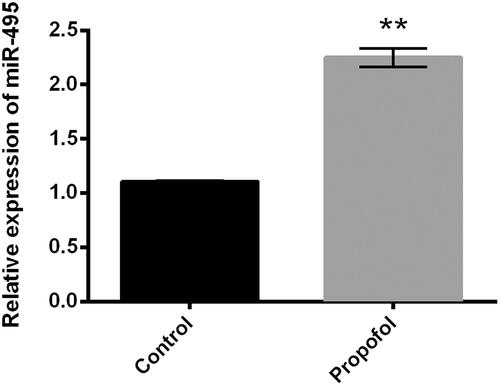
Suppression of miR-495 blocks the suppressing effects of propofol in cell proliferation and metastasis in JEG-3 cells
JEG-3 cells were transfected with miR-495 inhibitor and co-treated with propofol; the expression of miR-495 was detected using qRT-PCR. Transfection with miR-495 inhibitor significantly declined the miR-495 level, which indicated high transfection efficiency () (p < .01). Consistently, we found that the inhibitive effect of propofol on cell viability and metastasis was blocked by miR-495 inhibitor. miR-495 significantly increased cell viability (, p < .01), cell proliferation (), migration and invasion (, p < .05, or p < .01), and accelerated EMT process, as down-regulated E-cadherin and up-regulated N-cadherin and Vimentin levels (). Overall, these results indicated that propofol inhibited cell viability and metastasis and inhibited the EMT via up-regulation of miR-495.
Figure 3. Suppression of miR-495 blocks the regulation of propofol in cell proliferation and metastasis in JEG-3 cells. JEG-3 cells were transfected with miR-495 inhibitor and the corresponding negative control and then were treated with propofol (1 μM). (A) The expression of miR-495 was measured using qRT-PCR. Then, (B) cell viability, (C) cell proliferation, (D) relative migration and (E) invasion, and (F) the expression of EMT-regulated core factors were respectively detected using CCK-8 assay, Ki67+ assay, Transwell analysis, and western blot analysis. *p < .05, **p < .01.
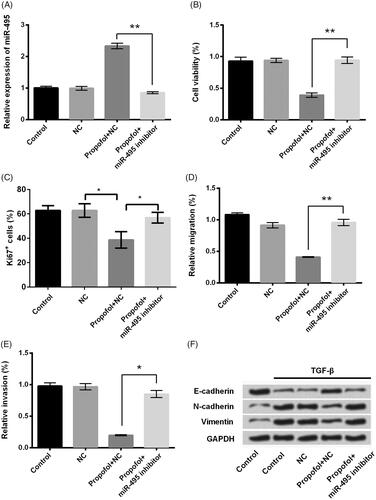
Propofol decreases Twist1 expression through up-regulating miR-495
Increasing evidence from previous studies revealed that miR-495 could regulate cell proliferation, migration and invasion via targeting AKT in prostate cancer cells [Citation22], in oesophagal squamous cell carcinoma [Citation8], and colorectal cancer [Citation23]. Obviously, there is a close correlation between miR-495 and AKT pathway. Motivated by this information, we also detected the effects of miR-495 on the expression of AKT1 and result showed that miR-495 downregulation increased the expression of AKT1 which reversed the result induced by propofol (p < .05, ). This result indicated that miR-495 also influence the expression of AKT1 as what the previous studies have reported. This result illustrated that miR-495 and AKT might be an explanation about the mechanism. However, we were looking forward to investigating something different; then, we surprisingly found that Twist1 had been reported as an up-stream factor promoting the process of EMT [Citation24]. Therefore, we intended to know whether Twist1 was involved in the progression of propofol inhibited EMT. Then, we investigated the effects of propofol on Twist1 expression using qRT-PCR and western blot analysis. As shown in , the mRNA and protein expression of Twist1 was significantly reduced in JEG-3 cells after propofol treatment (p < .01). However, miR-495 inhibitor markedly reversed the effect of propofol and increased the JEG-3 expression (p < .01). These data revealed that propofol down-regulates Twist1 expression via up-regulation of miR-495. It is hard to exclude the effects of AKT because mechanisms about the effects of miR-495 on this cancer are extremely complex. Twist 1 can only be treated as one of the explanations, but there might possibly be other mechanisms that responsible for these effects.
Figure 4. Propofol decreases AKT1 and Twist1 expression through regulating miR-495. JEG-3 cells were transfected with miR-495 inhibitor and the corresponding negative control and then were treated with propofol (1 μM). Then, the mRNA and protein expression of (A) AKT1 and (B) Twist1 was evaluated using qRT-PCR and western blot analysis. *p < .05, **p < .01.
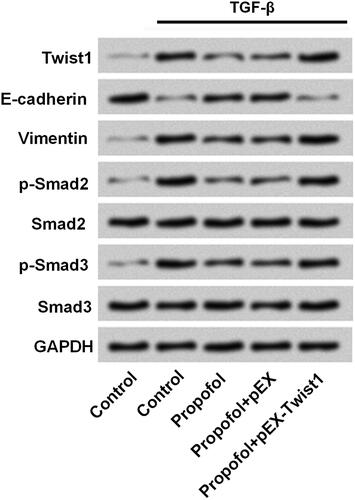
Twist1 participates in the regulation of propofol on TGF-β signalling inactivation in JEG-3 cells
As aforementioned, we found that propfofol could influence the expression of Twist1, which might through regulation of Twist1 to affect TGF-β signalling. In addition, amount of report showed that Smad proteins as pivotal intracellular effectors of TGF-β1 [Citation25]; hence, we explored the effects of Twist1 on TGF-β/Smad signalling pathway. As shown in , the TGF-β signalling was inactivated by propofol, as a reduce levels of Vimentin, p-Smad2 and p-Smad3, and an increase of E-cadherin. However, overexpression of Twist1 reversed the effect of propofol on these factors. It suggested that propofol regulated TGF-β/Smad signalling process through down-regulation of Twist1.
Figure 5. Twist1 participates in the regulation of propofol on TGF-β signalling inactivation in JEG-3 cells. JEG-3 cells were transfected with Twist1 overexpress-vector and the corresponding negative control. The protein expressions of Twist1, E-cadherin, Vimentin, p-Smad2, Smad2, p-Smad3 and Smad3 were measured by western blot.
Discussion
Propofol is widely used for induction or maintenance of anesthesia during certain surgeries, tests or procedures. Emerging evidences have demonstrated that propofol exerts an anti-tumour effect in various kinds of cancers including lung cancer, hepatocellular carcinoma, pancreatic cancer, ovarian cancer and breast cancer in the reduction of tumour size and weight [Citation26,Citation27], promotion of cancer cell apoptosis [Citation27,Citation28] and suppression of migration and invasion of cancer cells [Citation29,Citation30]. The effect of propofol in choriocarcinoma has not been elucidated yet.
In the current study, results revealed that propofol significantly blocked cell viability, inhibited migration and invasion, and EMT process in JEG-3 cells, which were consistent with results from the previous study of propofol in other cancers [Citation29–31].
Previous studies have demonstrated that propofol function as its regulation on cancer cell through miRNAs. For example, propofol inhibited hepatocellular carcinoma tumour growth by microvesicle-mediated transfer of miR-142-3p from macrophage to cancer cells [Citation26] and the adhesion by up-regulating miR-199a [Citation32]. Another report demonstrated that propofol-induced apoptosis of epithelial ovarian cancer cells through up-regulation of miRNA let-7i, and inhibition of miR-let-7i reversed the effect of propofol on cell proliferation and apoptosis [Citation28]. Propofol also suppressed proliferation and invasion of gastric cancer cells via down-regulation of miR-221 [Citation33]. Among all these identified miRNAs, the dysregulation of miR-495 has been reported in a variety of types of cancers, in which miR-495 was involved in the modulation of cell proliferation, migration and invasion, EMT process, and apoptosis [Citation8,Citation10,Citation12,Citation34]. To clarify the mechanism underlying in the suppression of JEG-3 cells, the effect of propofol on miR-495 expression was examined. In the current study, we found that miR-495 expression was up-regulated by propofol in JEG-3 cells and miR-495 inhibition reversed the effect of propofol in JEG-3 cell viability, migration and invasion, and the process of EMT.
N-cadherin, Vimentin and Twist are the specific EMT markers [Citation35]. Suppression of E-cadherin transcription and promotion of N-cadherin, Vimentin and fibronectin play important role in human breast epithelial cells [Citation36]. The previous study revealed that EMT could function in promoting carcinoma invasion and metastasis [Citation37]. Our study showed that propofol could inhibit EMT, which lined up with the results that propofol inhibited cell viability and metastasis.
Furthermore, previous literature reported that Twist1 could also induce EMT [Citation38]. Moreover, Twist1 is a key downstream effector of p62 in the regulation of cell proliferation and migration [Citation39]. In our study, we demonstrated that the expression of Twist1 was down-regulated by propofol through up-regulation of miR-495. Then, we asked whether there was connection between TGF-β signalling with Twists1 since both of them could induce EMT. Afterwards, we further investigated whether propofol function as its modulation on EMT process through regulating Twist1. TGF-β signalling pathway plays an significant and albeit complex role in the progression of tumourigenesis, which closely linked to EMT, apoptosis and proliferation in cancer cells [Citation40,Citation41]. A key pathway that correlated with the regulation of TGF-β induced effects is the canonical Smad pathway [Citation25]. We found that propofol inhibited the activation of TGF-β/Smad pathway, as evidenced by increasing E-cadherin, and decreasing Vimentin, p-Smad2 and p-Smad3. However, overexpression of Twist1 antagonizes the effect of propofol on TGF-β/Smad pathway inactivation. It suggested that Twist1 was required in the regulation of propofol on TGF-β signalling, and it might be one of mechanisms that propofol exerts its anti-tumour function in JEG-3 cells.
In conclusion, the present study demonstrated that propofol inhibited cell proliferation, migration and invasion, and EMT process in JEG-3 cells. And, the mechanism might be through up-regulation of miR-495 and down-regulation of Twist1. To the best of our knowledge, this is the first study to demonstrate the effect of propofol on choriocarcinoma, and it may provide new insight for the treatment of choriocarcinoma in the future. Further studies are suggested to clear the exact effect of propofol on choriocarcinoma in animal models and in the clinic.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Kurman RJ, Carcangiu ML, Herrington CS. WHO classification of tumours of female reproductive organs. Lyon: International Agency for Research on Cancer (IARC); 2014.
- Hertz R, Mc LI, Spencer DB. Effect of methotrexate therapy upon choriocarcinoma and chorioadenoma. Proc Soc Exp Biol Med. 1956;93:361.
- Lurain JR. Gestational trophoblastic disease I: epidemiology, pathology, clinical presentation and diagnosis of gestational trophoblastic disease, and management of hydatidiform mole. Am J Obstet Gynecol. 2010;203:531–539.
- Chao A, Tsai CL, Wei PC, et al. Decreased expression of microRNA-199b increases protein levels of SET (protein phosphatase 2A inhibitor) in human choriocarcinoma. Cancer Lett. 2010;291:99–107.
- Iem S. Gestational trophoblastic neoplasia–pathogenesis and potential therapeutic targets. Lancet Oncol. 2007;8:642.
- Bartel D. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297.
- Chen CZ. MicroRNAs as oncogenes and tumor suppressors. N Engl J Med. 2005;302:1–12.
- Mao Y, Li L, Liu J, et al. MiR-495 inhibits esophageal squamous cell carcinoma progression by targeting Akt1. Oncotarget. 2016;7:51223–51236.
- Hee LS, Dong JY, Sun CY, et al. Targeting of RUNX3 by miR-130a and miR-495 cooperatively increases cell proliferation and tumor angiogenesis in gastric cancer cells. Oncotarget. 2015;6:33269–33278.
- Hwang-Verslues WW, Chang P-H, Wei P-C, et al. miR-495 is upregulated by E12/E47 in breast cancer stem cells, and promotes oncogenesis and hypoxia resistance via downregulation of E-cadherin and REDD1. Oncogene. 2011;30:2463.
- Cao M, Nie W, Li J, et al. MicroRNA-495 induces breast cancer cell migration by targeting JAM-A. Protein Cell. 2014;5:862–872.
- Xu YY, Jing T, Quan H, et al. MicroRNA-495 downregulates FOXC1 expression to suppress cell growth and migration in endometrial cancer. Tumor Biol. 2016;37:239–251.
- Li Z, Cao Y, Jie Z, et al. miR-495 and miR-551a inhibit the migration and invasion of human gastric cancer cells by directly interacting with PRL-3. Cancer Lett. 2012;323:41–47.
- Miyata T, Kodama T, Honma R, et al. Influence of general anesthesia with isoflurane following propofol-induction on natural killer cell cytotoxic activities of peripheral blood lymphocytes in dogs. J Veter Med Sci. 2013;75:917.
- Altenburg JD, Harvey KA, Mccray S, et al. A novel 2,6-diisopropylphenyl-docosahexaenoamide conjugate induces apoptosis in T cell acute lymphoblastic leukemia cell lines. Biochem Biophys Res Commun. 2011;411:427–432.
- Mammoto T, Mukai M, Mammoto A, et al. Intravenous anesthetic, propofol inhibits invasion of cancer cells. Cancer Lett. 2002;184:165.
- Ye Z, Jingzhong L, Yangbo L, et al. Propofol inhibits proliferation and invasion of osteosarcoma cells by regulation of microRNA-143 expression. Oncol Res. 2013;21:201–207.
- Zhang J, Wu GQ, Zhang Y, et al. Propofol induces apoptosis of hepatocellular carcinoma cells by upregulation of microRNA-199a expression. Cell Biol Int. 2013;37:227–232.
- Inada T, Kubo K, Shingu K. Possible link between cyclooxygenase-inhibiting and antitumor properties of propofol. J Anesth. 2011;25:569–575.
- Huang Z, Li S, Fan W, et al. Transforming growth factor β1 promotes invasion of human JEG-3 trophoblast cells via TGF-β/Smad3 signaling pathway. Oncotarget. 2017;8:33560–33570.
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829.
- Li JZ, Wang ZL, Xu WH, et al. MicroRNA-495 regulates migration and invasion in prostate cancer cells via targeting Akt and mTOR signaling. Cancer Invest. 2016;34:181–188.
- Yan L, Yao J, Qiu J. miRNA-495 suppresses proliferation and migration of colorectal cancer cells by targeting FAM83D. Biomed Pharmacother. 2017;96:974–981.
- Meng J, Chen S, Han JX, et al. Twist1 regulates vimentin through Cul2 circular RNA to promote EMT in hepatocellular carcinoma. Cancer Res. 2018;78:4150–4162.
- D'Arpino MC, Fuchs AG, Sanchez SS, et al. Extracellular matrix remodeling and TGF-beta1/Smad signaling in diabetic colon mucosa. Cell Biol Int. 2018;42:443–456.
- Zhang J, Shan WF, Jin TT, et al. Propofol exerts anti-hepatocellular carcinoma by microvesicle-mediated transfer of miR-142-3p from macrophage to cancer cells. J Transl Med. 2014;12:1–9.
- Cui WY, Liu Y, Zhu YQ, et al. Propofol induces endoplasmic reticulum (ER) stress and apoptosis in lung cancer cell H460. Tumour Biol. 2014;35:5213–5217.
- Z S, XK H, QP W. Propofol induces apoptosis of epithelial ovarian cancer cells by upregulation of microRNA let-7i expression. Eur J Gynaecol Oncol. 2014;35:688.
- Wang P, Chen J, Mu LH, et al. Propofol inhibits invasion and enhances paclitaxel- induced apoptosis in ovarian cancer cells through the suppression of the transcription factor slug. Eur Rev Med Pharmacol Sci. 2013;17:1722–1729.
- Xu YB, Du QH, Zhang MY, et al. Propofol suppresses proliferation, invasion and angiogenesis by down-regulating ERK-VEGF/MMP-9 signaling in Eca-109 esophageal squamous cell carcinoma cells. Eur Rev Med Pharmacol Sci 2013;17:2486.
- Wu KC, Yang ST, Hsia TC, et al. Suppression of cell invasion and migration by propofol are involved in down-regulating matrix metalloproteinase-2 and p38 MAPK signaling in A549 human lung adenocarcinoma epithelial cells. Anticancer Res. 2012;32:4833–4842.
- Zhang J, Zhang D, Wu G-Q, et al. Propofol inhibits the adhesion of hepatocellular carcinoma cells by upregulating microRNA-199a and downregulating MMP-9 expression. Hepatobiliary Pancreat Dis Int. 2013;12:305–309.
- Wang ZT, Gong HY, Zheng F, et al. Propofol suppresses proliferation and invasion of gastric cancer cells via downregulation of microRNA-221 expression. Genet Mol Res. 2015;14:8117.
- Wei T, Zhu W, Fang S, et al. miR-495 promotes the chemoresistance of SCLC through the epithelial-mesenchymal transition via Etk/BMX. Am J Cancer Res. 2017;7:628.
- Martin FT, Dwyer RM, Kelly J, et al. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: stimulation of epithelial to mesenchymal transition (EMT). Breast Cancer Res Treat. 2010;124:317–326.
- D'Angelo RC, Liu XW, Najy AJ, et al. TIMP-1 via TWIST1 induces EMT phenotypes in human breast epithelial cells. Mol Cancer Res. 2014;12:1324–1333.
- Zhang P, Sun Y, Ma L. ZEB1: at the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle. 2015;14:481–487.
- Casas E, Kim J, Bendesky A, et al. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 2011;1;71:245–254.
- Qiang L, Zhao B, Ming M, et al. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc Natl Acad Sci USA. 2014;111:9241–9246.
- Yu JR, Tai Y, Jin Y, et al. TGF-β/Smad signaling through DOCK4 facilitates lung adenocarcinoma metastasis. Genes Dev. 2015;29:250–261.
- Heldin C-H, Landström M, Moustakas A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol. 2009;21:166–176.