
Abstract
Atherosclerosis is the chronic inflammatory disease, and inflammation-elicited endothelial activation is an early event in the development of atherosclerosis. The P2Y11 receptor is a purinergic receptor and a member of the P2 family of G coupled protein which has been shown to modulate vascular function. Progress in the study of purine receptors has been tremendous and these receptors have become pharmacological targets for various diseases. In this study, we show that the P2Y11R antagonist NF157 can mitigate oxidized LDL (ox-LDL)-induced endothelial inflammation. Our study demonstrates that P2Y11R is expressed to a fair degree in human aortic endothelial cells and is induced by treatment with ox-LDL. Blockage of P2Y11R by its selective antagonist NF157 ameliorates ox-LDL-induced adhesion of THP-1 monocytes to endothelial cells. NF157 inhibits ox-LDL-induced expression of adhesion molecules including E-selectin and VCAM-1. NF157 also suppresses ox-LDL-associated ROS production and induction of the NADPH oxidase subunit NOX-4. Moreover, NF157 has an inhibitory effect on the production of major cytokines including IL-6 and TNF-α. Mechanistically, we show that NF157 mitigates ox-LDL-induced phosphorylation of MAPK kinase p38 and NF-κB activation. Our findings indicate that blockage of P2Y11R signalling by its antagonist NF157 may protect endothelial cells from ox-LDL-induced endothelial inflammation. Therefore, NF157 may have therapeutic implications in the modulation of atherosclerosis-associated inflammation.
Introduction
Cardiovascular disease is a major contributor to mortality and morbidity in many countries. Many cardiovascular diseases are caused by atherosclerosis. Atherosclerosis originates from the buildup of fatty plaque on the artery walls and eventually results in narrowing and clogging of the vessel walls. Located on the inner layer of the blood vessels, endothelial cells interconnect to ensure sufficient supply of blood and availability of nutrients for every organ in the body. Due to the large number of blood vessels and their huge surface area, the endothelium is the largest organ in the body, so its normal function is critical to maintaining a balanced state of whole-body hemostasis [Citation1]. Under normal conditions, endothelial cells exist in a quiescent state and their functions are activated by blood flow and various physiological stimuli in the circulation. However, activation of endothelial cells and other harmful stimuli can lead to endothelial dysfunction, which is now recognized as an early event in the development of atherosclerosis [Citation2]. One of the most deleterious stimuli to vascular endothelial cells comes from the oxidation of LDL (ox-LDL). Ox-LDL is an important risk factor contributing to endothelial dysfunction [Citation3]. Ox-LDL exerts critical actions within the vascular endothelial microenvironment and influences leukocyte adhesion, platelet aggregation, and vascular smooth-muscle cell proliferation and migration. Ox-LDL is regarded as an oxidative stress biomarker and one of the primary risk factors for atherosclerosis [Citation4].
Extracellular ATP as well as other purine and pyrimidine nucleotides not only act as major energy carriers in biological systems but also function as important extracellular signalling molecules. ATP and other nucleotide signals have been found to bind to the membrane-bound purine receptors and mediate extracellular signalling through these receptors [Citation5]. There are two main families of purine receptors: P1 and P2, which are membrane-bound G-proteins or G-protein-coupled receptors. There are two types of P2 receptors: G-protein-coupled P2Y receptors and ATP-activated ligand-gated ion channel P2X receptors. Purinergic signalling plays important roles in controlling vascular tone and vessel remodelling [Citation6]. Two purine receptors including the P2Y1 and P2Y11 receptors are the most abundantly expressed P2Y receptors in the endothelium [Citation7]. In our study of endothelial activation in atherosclerosis, we found that P2Y11R is a highly specific inducible factor which response to ox-LDL stimulation. The present study evaluates the effect of P2Y11R in endothelial cells.
Materials and methods
Cell culture, adhesion and treatment experiment
Human primary aortic endothelial cells (HAECs) were purchased from Lonza (CC-2635). HAECs were grown in 2% serum endothelial growth media (EGM2) in low passage numbers (less than 10). Human monocyte cell line THP-1 cells were from ATCC stock (TIB-202™) and grown in 1640 medium supplemented with 10% fetal bovine serum (FBS). Human huh7 hepatocellular carcinoma was purchased from ATCC and maintained in DMEM growth media supplemented with 10% FBS. For ox-LDL treatment, 100 mg/mL freshly prepared ox-LDL-containing growth media was added to confluent HAECs media for 24 or 48 h depending on the experiment. For blockage of P2Y11R co-treatment experiments, 25 and 50 μM NF157 was added to confluent HAECs media for 24 h. For cell adhesion experiments, suspension growing THP-1 cells were pre-labelled with the dye calcein-AM. Cell adhesion experiments were performed by mixing 5 × 106 THP-1 cells with 1 × 106 confluent HAECs for 1 h. Adhesive THP-1 cells on the endothelial surface were washed and visualized using a fluorescence microscope.
Quantitative real-time PCR analysis
RNA was isolated from HAECs using an RNeasy Micro Kit from Qiagen (Cat.74004). 1 μg RNA was used for a reverse transcription PCR (RT-PCR) to produce cDNA using iScript Reverse Transcription Supermix (Bio-Rad, #1708840). The mRNA levels of human P2Y11R, NOX-4, E-selectin, VCAM-1, IL-6, and TNF-α were assessed by real-time PCR with SYBR Green master mix on a 7500 Real-time PCR platform as previously described [Citation8].
Western blot analysis
Protein was extracted from HAECs using RIPA buffer with protease and phosphatase inhibitors as previously described [Citation8]. The nuclear extracts from HAECs were prepared using a commercial kit (Thermo Fisher Scientific, USA). 20 μg protein extracts were separated by 10% SDS-PAGE and transferred to PVDF membranes. Membranes were incubated with specific antibodies and secondary antibodies. The immunoblots were developed with ECL substrate (Catalog #32132, Pierce). The following primary antibodies were used: P2Y11R, NOX-4, E-selectin, VCAM-1, p38, p-p38, p65, lamin B1 and β-actin.
ROS measurement
Cellular production of ROS was measured by quick staining the cells from the different conditions with DCFH-DA as previously described [Citation9]. After stimulation, cells were loaded with DCFH-DA (10 μM) for 30 min. The fluorescent image density was captured by a fluorescent microscope. Quantification of fluorescent density was performed using ImageJ software.
Enzyme-linked immunosorbent assay (ELISA)
To measure the secreted levels of IL-6 and TNF-α, culture media of HAECs was used for ELISA analysis with commercial ELISA kits from R&D Systems as reported previously [Citation9]. The quantification of these pro-inflammatory cytokines was performed by being normalized to protein concentration under different conditions.
Luciferase promoter assay
NF-κB luciferase vector was purchased from Thermo Fisher Scientific, USA. Cells were co-transfected with NF-κB promoter plus a firefly luciferase promoter using Lipofectamine 2000 (#11668027, Invitrogen, USA) as previously reported [Citation9]. 24 h later, cells were stimulated with ox-LDL (100 mg/L) with or without NF157 (25 and 50 μM) for another 24 h. Cell lysates were prepared to examine the luciferase activity.
Transcriptional activity of NF-κB was assessed by normalizing the luciferase activity of firefly to renilla.
Statistical analysis
Experimental data are shown as means ± standard derivation (SD). Comparisons among different groups were performed using the one-way ANOVA test, and p values less than 0.05 were determined to be statistically significant.
Results
P2Y11R is expressed in human aortic endothelial cells
First, we investigated whether P2Y11R is expressed in HAECs by detecting both the mRNA and protein levels of P2Y11R in primary HAECs with huh-7 cells as the reference cell line, which originated from hepatocellular cancer and are known to express a fair amount of P2Y11R [Citation10]. Compared to huh-7 cells, HAECs expressed a similar amount of P2Y11R mRNA () and protein (), and this expression profile laid the basis for our further investigation of the role of this receptor in endothelial cells.
Figure 1. P2Y11R is expressed on primary human aortic endothelial cells (HAECs). Human huh-7 cells were used as a positive control. (A) RT-PCR of P2Y11R; (B) Western blot analysis of P2Y11R.

Ox-LDL induces P2Y11R expression in endothelial cells
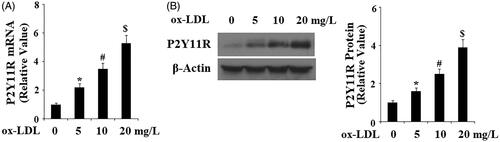
To reveal the role of P2Y11R, we challenged HAECs with the pathophysiologically relevant reagent ox-LDL. Ox-LDL is one of the major factors for the development of atherosclerosis and may lead to the activation of quiescent endothelial cells. Compared to non-treatment cells, P2Y11R mRNA rose gradually along with the increase in the concentration of ox-LDL, with 5, 10 and 20 mg/L of ox-LDL inducing approximately 2-, 3.5- and 5.5-fold expression of P2Y11R, respectively (). We were able to confirm this dose-dependent induction at the protein level. Again, compared to non-treatment cells, 5, 10 and 20 mg/L ox-LDL induced roughly 1.5-, 2.7- and 4-fold expression of P2Y11R protein, respectively ().
Figure 2. Ox-LDL increases the expression of endothelial P2Y11R in a dose-dependent manner. HAECs were treated with ox-LDL at the concentrations of 0, 5, 10 and 20 mg/L for 24 h. (A) Real-time PCR analysis of P2Y11R; (B) Western blot analysis of P2Y11R (*, #, $, p < .01 vs. vehicle group).
P2Y11R antagonist NF157 ameliorates ox-LDL-induced attachment of monocytes to endothelial cells
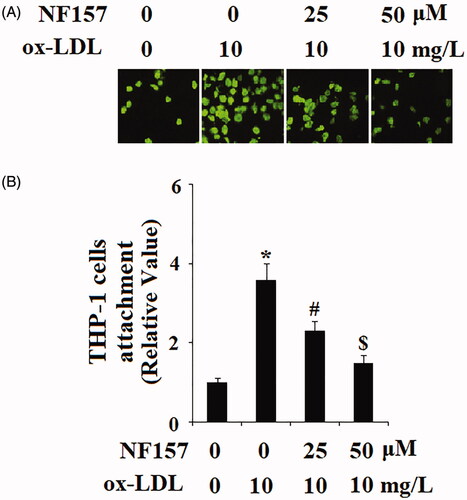
The inductive response of P2Y11R indicates that P2Y11R elevation resulted from ox-LDL-induced cellular stress and suggests that P2Y11R could be involved in reactions triggered by ox-LDL. For endothelial cells, an important function of P2Y11R is conditional immune protection to prevent immune cells from invading the vascular wall. However, under stress from ox-LDL, immune cells can break this protective barrier and attach to endothelial cells. We set out to block P2Y11R using its selective antagonist NF157 to test its influence on immune-endothelial cell interactions via adhesion assay. Compared with non-treated cells, ox-LDL gave rise to binding of roughly 3.7 times more THP-1 to HAECs. However, when the two doses (25 and 50 μM) of NF157 were added under these conditions, ox-LDL only resulted in binding of about 2.2 and 1.5 times more THP-1 cells to HAECs (). This experiment indicates that inactivation of P2Y11R may partly relieve ox-LDL-induced adhesion of immune cells to endothelial cells, and P2Y11R could be one of the mediators of ox-LDL-induced cellular stress.
Figure 3. The P2Y11R antagonist NF157 attenuates ox-LDL-induced adhesion of THP-1 monocytes to HAECs. HAECs were treated with 10 mg/L ox-LDL with or without NF157 (25, 50 μM) for 24 h. (A) Representative images of adhesion of THP-1 cells to HAECs; (B) Quantification of adhesive THP-1 cells (*, #, $, p < .01 vs. previous column group).
NF157 inhibits ox-LDL-induced expression of E-selectin and VCAM-1
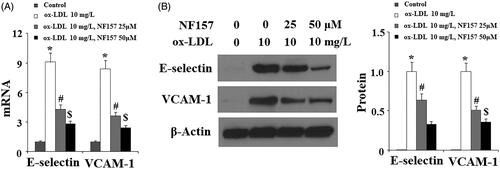
Ox-LDL is known to activate endothelial cells to release inflammatory cytokines and vascular adhesion molecules. We evaluated whether NF157 could affect the production of pro-inflammatory factors. Compared to non-treated cells, ox-LDL resulted in about nine-fold mRNA transcription of E-selectin while only causing about 4.5- and 2.5-fold higher E-selectin mRNA transcripts after blockade of P2Y11R using 25 and 50 μM NF157, respectively (). Similarly, ox-LDL caused approximately 8.5-fold higher mRNA transcripts of VCAM-1 while only triggering approximately 3.5- and 2.5-fold higher VCAM-1 after treatment with 25 and 50 μM NF157, respectively (). We further confirmed the inhibitory effect of NF157 on these factors at the protein level. Under non-treatment quiescent conditions, HAECs have negligible levels of E-selectin and VCAM-1 expression while their protein expressions were elevated upon ox-LDL treatment. Quantitated to ox-LDL treatment, 25 and 50 μM NF157 could mitigate E-selectin elevation by 20% and 65%, respectively (). Similarly, these two doses of NF157 were able to mitigate the induction of VCAM-1 by ox-LDL by 50% and 60% ().
Figure 4. The P2Y11R antagonist NF157 inhibits ox-LDL-induced expression of E-selectin and VCAM-1. HAECs were treated with 10 mg/L ox-LDL with or without NF157 (25, 50 μM) for 24 h. (A) Real-time PCR analysis of E-selectin and VCAM-1; (B). Western blot analysis of E-selectin and VCAM-1 (*, #, $, p < .01 vs. previous column group).
NF157 ameliorates ox-LDL-induced oxidative stress
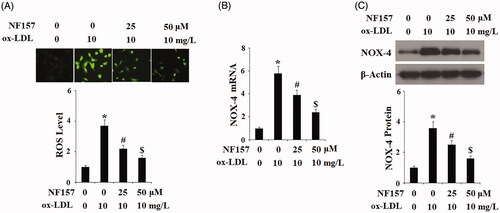
We then assessed whether blockage of P2Y11R had any influence on another aspect of ox-LDL-induced cellular stress response—oxidative stress. Two experiments were performed to assess ROS status: the level of total cellular ROS and NADPH oxidase. To measure the total cellular ROS level, cells were stained with DCFH-DA. As illustrated in the representative images and compared to the control, ox-LDL gave rise approximately 3.8-fold production of ROS. However, upon treatment with 25 and 50 μM NF157, the same concentration of ox-LDL caused only approximately 2- and 1.5-fold production of ROS, respectively (). NADPH oxidase is the major machinery of cellular ROS production, so we examined its major subunit NOX-4 in endothelial cells. At the mRNA level and compared to the control, ox-LDL resulted in 5.8-fold endothelial NOX-4 expression. However, in the presence of 25 and 50 μM NF157, the same concentration of ox-LDL caused only approximately 3.8- and 2.2-fold NOX-4 transcription (). We were able to confirm this suppression at the protein level. Compared to the control group, ox-LDL induced approximately 3.5-fold higher NOX-4 protein expression. However, in the presence of 25 and 50 μM NF157, the same concentration of ox-LDL caused only approximately 2.5- and 1.5-fold NOX-4 transcription (). These evidence indicate that blockage of P2Y11R by NF157 indeed ameliorated ox-LDL-induced endothelial oxidative stress.
Figure 5. NF157 inhibits ox-LDL-induced oxidative stress. HAECs were treated with 10 mg/L ox-LDL with or without NF157 (25, 50 μM) for 24 h. (A) Production of reactive oxygen species (ROS); (B) Expression of NOX-4 at the mRNA level; (C) Expression of NOX-4 at the protein level (*, #, $, p < .01 vs. previous column group).
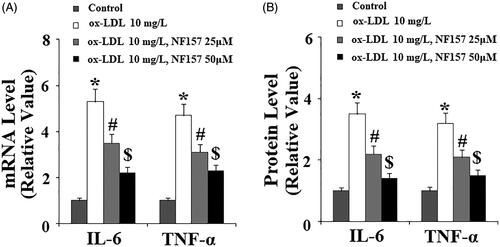
NF157 inhibited ox-LDL-induced IL-6 and TNF-α production
Next, we assessed the effect of NF157 on the production of pro-inflammatory cytokines. We monitored changes in the levels of both transcripted and secreted IL-6 and TNF-α. At the mRNA level and compared with non-treated cells, ox-LDL caused approximately 5.5-fold IL-6 transcription. However, there was only approximately 3.8- and 2.2-fold induction when the two doses of NF157 were present in the media, respectively (). Similarly, ox-LDL gave rise to approximately 5-fold TNF-α transcription, but only approximately 3.5- and 2.2-fold induction when the two doses of NF157 were added, respectively (). We confirmed these inhibitions at the protein level. Compared to the control, ox-LDL resulted in approximately 3.5-fold higher IL-6 expression, but only approximately 2.2- and 1.2-fold higher expression of IL-6 when the two doses NF157 were added to the media, respectively (). Similarly, ox-LDL caused approximately 3.3-fold higher TNF-α expression, but only approximately 2- and 1.5-fold induction when the two doses of NF157 were present (). Therefore, we concluded that NF157 had an inhibitory role in ox-LDL-induced induction of pro-inflammatory cytokines.
Figure 6. NF157 inhibits ox-LDL-induced expression of the pro-inflammatory cytokines IL-6 and TNF-α. HAECs were treated with 10 mg/L ox-LDL with or without NF157 (25, 50 μM) for 24 h. (A) Real-time PCR analysis of IL-6 and TNF-α at the mRNA level; (B) ELISA analysis of IL-6 and TNF-α at the protein level (*, #, $, p < .01 vs. previous column group).
NF157 has been identified as an antagonist of both P2Y11R and P2X1 receptor (P2X1R). Therefore, we examined whether the selective P2X1 antagonist NF449 has a similar protective effect against ox-LDL-induced endothelial insults. Results in Supplementary Figure 1 indicated that NF157 but not NF449 could inhibit ox-LDL-induced expression of IL-6, TNF-α, E-selectin, and VCAM-1, as well as the attachment of THP-1 monocytes to HAECs, suggesting that the inhibitory effect of NF-157 on ox-LDL-induced endothelial inflammation is dependent on the blockade of P2Y11R in endothelial cells, which is consistent with a previous study showing that NF157, but not NF449, partially suppressed LPS or ATP-induced IL-6 production from peritoneal macrophages [Citation11].
AR-C67085 is not an agonist of any other P2 receptor except P2Y11R. To further confirm the involvement of P2Y11R in the ox-LDL-induced endothelial inflammation, AR-C67085 was used. As expected, results in Supplementary Figure 2 indicate that treatment with AR-C67085 significantly exacerbated ox-LDL-induced expression of IL-6, TNF-α, E-selectin, and VCAM-1, as well as the attachment of THP-1 monocytes to HAECs.
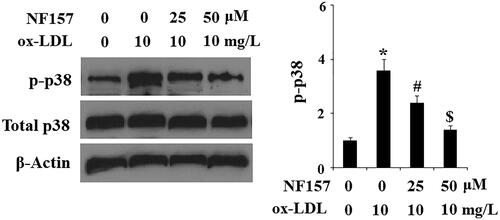
NF157 inhibits ox-LDL-induced activation of endothelial MAPK p38
Next, we explored the possible cellular signalling pathways involved in the protective effects of NF157 against ox-LDL-induced endothelial dysfunction. As the most disease-relevant stress inducer, ox-LDL is known to activate the endothelial MAPK p38 signalling pathway. We hypothesized that NF157 could interfere with endothelial MAPK p38 activation. Our dissemination of MAPK p38 and its phoryphation indeed confirmed this hypothesis. Compared with the control cells, ox-LDL caused approximately 3.8-fold higher p-p38 kinase. However, ox-LDL only induced approximately 2.2- and 1.5-fold higher p-p38 expression when 25 and 50 μM NF157 were present in the media, respectively. Meanwhile, ox-LDL and NF157 had no effect on total p38 expression ().
Figure 7. NF157 inhibits ox-LDL-induced activation of MAPK kinase p38. HAECs were treated with 10 mg/L ox-LDL with or without NF157 (25, 50 μM) for 2 h. Phosphorylated and total p38 were determined using western blot analysis (*, #, $, p < .01 vs. previous column group).
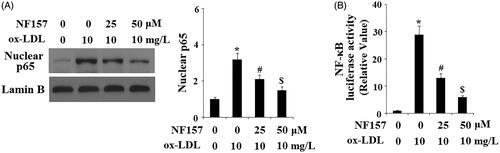
NF157 inhibits ox-LDL-induced activation of the endothelial NF-κB pathway
The p38-NF-κB axis pathway is a major inflammatory modulator in endothelial cells. Finally, we assessed the influence of NF157 on the NF-κB pathway. We monitored nuclear p65 accumulation and luciferase activity of NF-κB promoters. For nuclear p65, when compared to non-treated cells, ox-LDL resulted in accumulation of a large amount of p65, but this accumulation was gradually weakened by the presence of 25 and 50 μM NF157 (). For transfected NF-κB promoter and compared to non-treated cells, ox-LDL caused approximately 28-fold higher promoter activity, but only approximately 12- and 5-fold induction in the presence of the same two doses of NF157 (). These evidence demonstrate that NF157 suppresses the activation and transcription of both the p38 and NF-κB pathways.
Figure 8. NF157 inhibits ox-LDL-induced activation of NF-κB. HAECs were treated with 10 mg/L ox-LDL with or without NF157 (25, 50 μM) for 24 h. (A) Nuclear level of NF-κB protein p65. Lamin B1 was used as the loading control of nuclei; (B) NF-κB promoter luciferase activity change (*, #, $, p < .01 vs. previous column group).
Discussion
P2Y11R is a unique purinergic receptor among the members of the P2Y family as it couples to both the phosphoinositide and adenylyl cyclase pathways [Citation12]. Previous studies have identified P2Y11R as one of the most abundantly expressed P2Y receptors in the endothelium [Citation13]. Aside from its expression in endothelial cells, P2Y11R is also expressed on different immune cells and involved in immune cell movement, differentiation and other functions [Citation14]. P2Y11R has been characterized in different vascular endothelial cells. Previous studies have shown that extracellular ATP acts through aortic endothelial purinergic receptors including P2Y11R to regulate flow-induced vasodilatation [Citation15]. Endothelial P2Y11R has been shown to be a mediator in agonist-induced pulmonary endothelial cell barrier protection against lung injury [Citation7,Citation16]. Two studies have shown that P2Y11R is vital to endothelial cell survival and proliferation. Xiao et al. report that P2Y11R silencing in both HUVECs and HAECs by RNA interference reverses ATP-triggered inhibition of cell proliferation and P2Y11R deficiency ameliorates cell cycle arrest in the S phase [Citation17]. Helenius et al. show that P2Y11R-deficient pulmonary endothelial cells have a significant decrease in survival capacity, and several findings indicate that P2Y11 receptor signals could be involved in pulmonary hypertension [Citation18]. Aside from coupling to ATP and other nucleotide compounds, P2Y11R has been shown to be affinitive to nicotinamide adenine dinucleotide (NAD+) which is an important vascular mediator [Citation19]. One of the major advances in the P2Y11R field is the discovery of several specific P2Y11R antagonists including NF157 and NF340. NF157 has been identified as a potent inhibitor of P2Y11R activity [Citation20]. Several studies have shown that the P2Y11R-mediated response could be abolished by NF157 antagonist or P2RY11 knockdown [Citation16].
Our study aims to elucidate the role of P2Y11R in ox-LDL-induced endothelial inflammation as compounds targeting P2Y11R are readily available and highly selective, making this a promising treatment option. An appealing finding from our data confirms that P2Y11R is not only expressed in human aortic endothelial cells but is also responsive to ox-LDL treatment. The inductive nature of P2Y11R by ox-LDL suggests that this receptor may be a responsive mediator of vascular function. Thus, we adopted the available highly selective P2Y11R antagonist NF157 to block P2Y11R activity and conducted a series of experiments under treatment with ox-LDL. Our study provides four lines of evidence that P2Y11R blockage could be protective to vascular function. Firstly, blockage of P2Y11R by NF157 ameliorates ox-LDL-induced adhesion of monocytes to endothelial cells. This indicates that P2Y11R-mediated signalling is involved in cell adhesion elicited by ox-LDL and its inactivation could relieve the attachment of immune cells to the vascular wall. Second, we found that blockage of P2Y11R by NF157 potently suppresses ox-LDL-induced expression of adhesion molecules including E-selectin and VCAM-1. Both E-selectin and VCAM-1 have been detected on human atherosclerosis-prone endothelial cells and on the surface of fibrous and lipid-containing human plaques [Citation21]. The expression of VCAM-1 has been shown to be closely associated with the development of atherosclerosis [Citation22]. The inhibitory effect of NF157 on these vascular adhesion molecules implies that it has a vascular protective effect in the context of atherosclerosis. Third, our data show that NF157 suppresses the release of major pro-inflammatory cytokines such as IL-6 and TNF-α. Therefore, NF157 treatment could mitigate the inflammatory response triggered by ox-LDL. Fourth, NF157 treatment appears to mitigate endothelial oxidative stress triggered by ox-LDL. Additionally, we show that NF157 suppresses ox-LDL-induced upregulation of the NAPDH oxidase subunit NOX-4. Endothelial NOX-4 is a major catalytic component of the NAPDH complex and is known to contribute to vascular ROS production [Citation23]. Therefore, the inhibitory effect of NF157 on NOX-4 could be one mechanism of its anti-ROS property. Overall, the inhibitory effect of NF157 on the expression of pro-inflammatory vascular adhesion molecules and cytokines explains its ability to ameliorate adhesion of immune cells to endothelial cells. Mechanistically, we show that NF157 suppresses activation of MAPK kinase p38 and NF-κB. Previous studies have shown that ox-LDL and its endothelial receptor could activate these two signalling pathways [Citation24]. Although we have no evidence elucidating how NF157 mediates P2Y11R inactivation and inhibition of p38/NF-κB, evidence of its inhibitive effects on these signals are very solid. For MAPK kinase, we provide evidence that NF157 not only suppresses p38 kinase activation but also reduces its promoter activity. For NF-κB, our data confirm that NF157 strongly suppresses both nuclear accumulation of NF-κB p65 protein and its promoter activity.
In conclusion, endothelial P2Y11R signals are critical to the phenotypic function of endothelial cells. Our study demonstrates that active P2Y11R is required for the deleterious effects of ox-LDL on endothelial cells. Blockage of P2Y11R by its antagonist NF157 ameliorates endothelial inflammation caused by ox-LDL to a considerable degree. The protection of NF157 on endothelial activation occurs via inhibition of the MAPK kinase and NF-κB signalling pathways. Therefore, blockage of P2Y11R mediated by NF157 could have therapeutic implications in modulating vascular inflammation in the development of atherosclerosis.
Supplemental Material
Download ()Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Anggård EE. The endothelium-the body's largest endocrine gland? J Endocrinol. 1990;127:371–375.
- Gimbrone MA, Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–636.
- Steinberg D, Parthasarathy S, Carew TE, et al. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 1989;320:915–924.
- Di Pietro N, Formoso G, Pandolfi A. Physiology and pathophysiology of oxLDL uptake by vascular wall cells in atherosclerosis. Vascul Pharmacol. 2016;84:1–7.
- Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–492.
- Burnstock G, Ralevic V. Purinergic signaling and blood vessels in health and disease. Pharmacol Rev. 2014;66:102–192.
- Umapathy NS, Zemskov EA, Gonzales J, et al. Extracellular beta-nicotinamide adenine dinucleotide (beta-NAD) promotes the endothelial cell barrier integrity via PKA- and EPAC1/Rac1-dependent actin cytoskeleton rearrangement. J Cell Physiol. 2010;223:215–223.
- Zhou X, Cai J, Liu W, et al. Cysteinyl leukotriene receptor type 1 (CysLT1R) antagonist zafirlukast protects against TNF-α-induced endothelial inflammation. Biomed Pharmacother. 2019;111:452–459.
- Ma S, Bai Z, Wu H, et al. The DPP-4 inhibitor saxagliptin ameliorates ox-LDL-induced endothelial dysfunction by regulating AP-1 and NF-κB. Eur J Pharmacol. 2019;851:186–193.
- Khalid M, Brisson L, Tariq M, et al. Carcinoma-specific expression of P2Y11 receptor and its contribution in ATP-induced purinergic signalling and cell migration in human hepatocellular carcinoma cells. Oncotarget. 2017;8:37278–37290.
- Sakaki H, Tsukimoto M, Harada H, et al. Autocrine regulation of macrophage activation via exocytosis of ATP and activation of P2Y11 receptor. PLoS One. 2013;8:e59778.
- Communi D, Govaerts C, Parmentier M, et al. Cloning of a human purinergic P2Y receptor coupled to phospholipase C and adenylyl cyclase. J Biol Chem. 1997;272:31969–31973.
- Wang L, Karlsson L, Moses S, et al. P2 receptor expression profiles in human vascular smooth muscle and endothelial cells. J Cardiovasc Pharmacol. 2002;40:841–853.
- Dreisig K, Kornum BR. A critical look at the function of the P2Y11 receptor. Purinergic Signal. 2016;12:427–437.
- Liu C, Mather S, Huang Y, et al. Extracellular ATP facilitates flow-induced vasodilatation in rat small mesenteric arteries. Am J Physiol Heart Circ Physiol. 2004;286:H1688–H1695.
- Zemskov E, Lucas R, Verin AD, et al. P2Y receptors as regulators of lung endothelial barrier integrity. J Cardiovasc Dis Res. 2011;2:14–22.
- Xiao Z, Yang M, Lv Q, et al. P2Y11 impairs cell proliferation by induction of cell cycle arrest and sensitizes endothelial cells to cisplatin-induced cell death. J Cell Biochem. 2011;112:2257–2265.
- Helenius MH, Vattulainen S, Orcholski M, et al. Suppression of endothelial CD39/ENTPD1 is associated with pulmonary vascular remodeling in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2015;308:L1046–L1057.
- Moreschi I, Bruzzone S, Nicholas RA, et al. Extracellular NAD + is an agonist of the human P2Y11 purinergic receptor in human granulocytes. J Biol Chem. 2006;281:31419–31429.
- Ullmann H, Meis S, Hongwiset D, et al. Synthesis and structure-activity relationships of suramin-derived P2Y11 receptor antagonists with nanomolar potency. J Med Chem. 2005;48:7040–7048.
- Galkina E, Ley K. Vascular adhesion molecules in atherosclerosis. Atvb. 2007;27:2292–22301.
- Cybulsky MI, Iiyama K, Li H, et al. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–1262.
- Ago T, Kitazono T, Ooboshi H, et al. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation. 2004; 109:227–233.
- Pirillo A, Reduzzi A, Ferri N, et al. Upregulation of lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) by 15-lipoxygenase-modified LDL in endothelial cells. Atherosclerosis. 2011; 214:331–337.