
Abstract
Hepatitis B virus is one of the main causes of hepatitis and hepatocellular carcinoma (HCC). Hepatitis B virus-encoded X protein (HBx) has been shown to be involved in many aspects of the pathogenicity of liver diseases. Orexin A is a small peptide produced in the hippocampus. Orexin A and its receptor have become important therapeutic targets for certain metabolic disorders. In this study, we show that orexin A has a protective role against HBx-induced cytotoxicity and inflammation in hepatocytes. The ectopic expression of HBx in hepatocytes reduces orexin A receptor 1 (OX1R) expression. When orexin A is added to the cells, it mitigates HBx-induced oxidative stress indicator 4-hydroxynonenal (4-HNE) and reactive oxygen species (ROS) as well the NADPH subunit NADPH oxidase 4 (NOX-4). Orexin A also ameliorates HBx-mediated mitochondrial membrane potential and adenosine triphosphate (ATP) reduction. Moreover, orexin A significantly inhibits HBx-induced production of pro-inflammatory cytokines including interleukin 8 (IL-8), tumour necrosis factor α (TNF-α) and chemokine ligand 2 (CXCL2). The presence of orexin A ameliorates HBx-induced lactate dehydrogenase (LDH) release, indicating that it could protect hepatocytes from cytotoxicity. Mechanistically, we found that orexin A suppresses c-Jun N-terminal kinase (JNK) phosphorylation, accumulation of nuclear factor-κB (NF-κB) protein p65 in nuclei, and NF-κB promoter activity, suggesting that orexin A suppresses JNK and NF-κB pathway activation. In conclusion, our study demonstrates that orexin A peptide possesses a protective role against HBx-mediated cytotoxicity and inflammation in hepatocytes.
Introduction
Hepatitis B virus (HBV) infection is a global public health problem that affects more than 300 million people worldwide and is a direct cause of hepatitis B [Citation1]. Chronic HBV infection also leads to liver cirrhosis and hepatocellular carcinoma, which are major contributors to the high prevalence of HBV infection-related mortality [Citation2]. The treatment of HBV infection remains a challenge. There is no specific treatment for acute hepatitis B. For chronic hepatitis B infection, treatment strategies rely on antiviral agents to slow the progress of cirrhosis and the occurrence of liver cancer. However, in most cases, antiviral agents are unable to cure hepatitis B infection due to the persistence of virus DNA in hepatocytes, so these treatments only suppress the replication rate of virus particles [Citation3].
HBV Infection of the liver may be either transient, chronic, or even lifelong, depending on the ability of the host immune response to clear the infection. Chronic infections can cause host immune-mediated liver damage, which can progress to cirrhosis and hepatocellular carcinoma. Extensive efforts have been made to prevent HBV infection-induced liver damage and host immune reactions before the disease can progress. The genome of HBV consists of four overlapped open reading frames (S, C, P and X) and expresses seven viral proteins. Among these viral proteins, HBx is one of the key determinants of viral replication and host infection. HBx is a small viral protein composed of 154 aa and can activate numerous inflammatory pathways and host-specific transcription factors [Citation4].
Many studies have focused on targeting HBx to prevent insult to host hepatocytes. The hypothalamic orexin A is a small neuropeptide which influences diverse physiological processes, such as control of food intake, sleep-wake behaviour, arousal, energy balance, and energy expenditure. Orexin A also activates cellular signals by binding its two target receptors, the orexin-1 receptor (OX1R) and the orexin-2 receptor (OX2R), both of which are G protein-coupled receptors (GCPRs) [Citation5]. Orexin A and its target receptors have become therapeutic targets for certain eating and sleeping disorders [Citation6]. While screening the potential host targets of HBx, we found that the introduction of HBx in hepatocytes reduced the expression of OX1R. Rescue experiments were performed and orexin A ligand was added to activate the receptor. Here, we report evidence substantiating the protective effects of orexin A against HBx-induced hepatocyte injury.
Materials and methods
Cell culture and treatment
Normal human hepatic cell line L-02 and MIHA were purchased from the American Type Culture Collection (ATCC). Cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) with 0.1% penicillin and streptomycin. For HBx expression and treatment experiments, 50% confluent L-02 and MIHA cells were transfected with HBx expression plasmids using Lipofectamine 2000 (Invitrogen). Twenty-four hours later, the cells were treated with 100 and 300 nM orexin A for another 24 h.
Real-time polymerase chain reaction (PCR) analysis
Total RNA from L-02 cells was isolated and purified using a High Pure RNA kit from Roche. Purified RNA (1 µg) from the different treatment groups was used to perform reverse transcription PCR (RT-PCR) to generate cDNA using an iScript Supermix kit (Invitrogen, USA). cDNA (2 μL) was used for subsequent real-time PCR analysis using an SYBR Green Master Mix kit (Roche) in a 20 μL reaction system to measure the gene expressions of OX1R, IL-8, TNF-α, CXCL2, NOX-4 and GAPDH on an ABI 7500 platform.
Western blot analysis
Isolated proteins 20 µg from the different groups were separated by polyacrylamide gel electrophoresis (PAGE). Samples were then electronically transferred to polyvinylidene fluoride (PVDF) membranes and incubated with their specific antibodies and corresponding secondary antibodies. The immunoblots were visualized using an enhanced chemiluminescence kit from Sigma-Aldrich, USA. The primary antibodies we used were OX1R, NOX-4, HBx, JNK, p-JNK, p65, Lamin B and β-actin.
Nuclear extracts
Nuclear proteins of L-02 cells were extracted using a kit from Thermo Fisher Scientific, USA. Nuclear extracts were used for western blot analysis with lamin B1 as quality control.
Enzyme-linked immunosorbent assay (ELISA)
Secretions of IL-8, TNF-α, and CXCL2 in L-02 and MIHA cells were measured by ELISA using commercial kits purchased from R&D Systems. Cell culture medium (100 μL) was collected and added to 96-well plates, then incubated overnight at 4 °C. Samples were then sequentially incubated with detection antibody, secondary antibody, stabilized chromogen and stop solution. The OD value at 450 nm was used to index secretions of IL-8, TNF-α and CXCL2.
Release of lactate dehydrogenase (LDH)
After the necessary transfection and stimulation for the time periods indicated, LDH released from L-02 cells was examined using a commercial kit (11644793001) from Sigma-Aldrich, USA. Briefly, 100 μL cell culture medium was harvested and mixed with 100 μL reaction solution. After incubation for 30 min, the OD value at 450 nm was used to index LDH release.
Mitochondrial membrane potential (MMP) measurement
MMP in L-02 cells was measured by staining cells with the dye tetramethyl rhodamine methyl and ethyl esters (TMRM) (Thermo Fisher Scientific, USA). After the necessary transfection and treatment, L-02 cells were loaded with 250 nM TMRM and incubated for 1 h at 37 °C. Cells were washed three times with PBS and red fluorescence was visualized using a fluorescence microscope. Fluorescent density was quantified using Image J to index the levels of MMP.
Determination of ROS
Cellular ROS was measured by staining L-02 cells with the ROS sensitive dye DCFH-DA (Sigma-Aldrich, USA). After the necessary transfection and treatment, L-02 cells were loaded with 1 μM DCFH-DA and incubated for 30 min at 37 °C. Cells were washed three times with PBS and green fluorescence was visualized using a fluorescence microscope. Fluorescence density was quantified using Image J software to index the levels of ROS.
Immunostaining of 4-hydroxynonenal (4-HNE)
The pattern of lipid peroxidation in L-02 cells was assessed by measuring the level of 4-HNE with immunostaining. After the necessary transfection and stimulation, cells were fixed with 4% paraformaldehyde and permeabilized using 0.1% Triton-x 100. Then, cells were sequentially probed with anti-HNE antibody and Alexa 488 conjugated secondary antibody. Green fluorescence was visualized using a fluorescence microscope. Fluorescence density was quantified using Image J software to index the levels of 4-HNE.
Intracellular ATP measurement
Intracellular levels of ATP in L-02 cells were assessed using a bioluminescent assay kit (Sigma-Aldrich, USA). After the necessary transfection and stimulation, cells were lysed. Cell lysates (50 μL) were mixed with equal volumes of reaction solution. Luminescence was measured using a single sample luminometer.
Promoter assay
L-02 and MIHA cells were co-transfected with NF-κB promoter and a firefly luciferase promoter using Lipofectamine 2000 (Invitrogen, USA). The transfected cells were pre-incubated with 100 and 300 nM orexin A for 24 h. The total cell lysates were prepared to examine the dual luciferase activity of renilla and firefly. The relative luciferase was calculated to reflect the promoter activity of NF-κB.
Statistical analysis
Data in this study are expressed as means ± SEM Statistical significance was tested using the ANOVA test. p < .05 was considered statistically significant.
Results
Overexpression of HBx reduces OX1R in hepatocytes
To map the responsive genes of HBx expression, we generated a mammalian HBx expression construct. We then transfected HBx plasmids into L-02 hepatocytes and verified its expression (). Next, we examined the influence of overexpression of HBx on hepatic OX1R. Our experiment showed that L-02 cells transfected with HBx produced roughly half the amount of OX1R transcripts and protein when compared to vehicle cells (). Clearly, HBx displays an inhibitory role on hepatic OX1R expression.
Figure 1. Determination of HBx expression in hepatocytes. HBx-encoding plasmids were transfected into L-02 hepatocytes for 48 h. Western blot analysis revealed the overexpression of HBx in normal human L-02 hepatocytes.
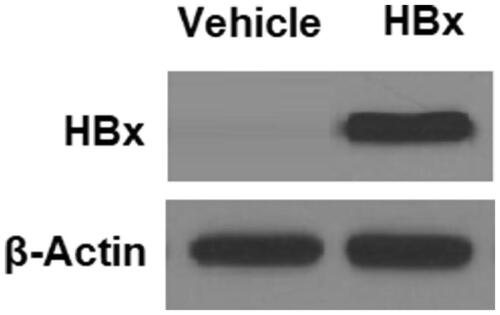
Figure 2. HBx reduces the expression of orexin-1 receptor (OX1R) in L-02 hepatocytes. The HBx-encoding plasmids were transfected into L-02 hepatocytes for 48 h. (A) Real-time PCR analysis revealed that overexpression of HBx decreased the expression of OX1R at the mRNA level; (B) Western blot analysis revealed that overexpression of HBx reduced the expression of OX1R at the protein level (*p < .01 vs vehicle group).
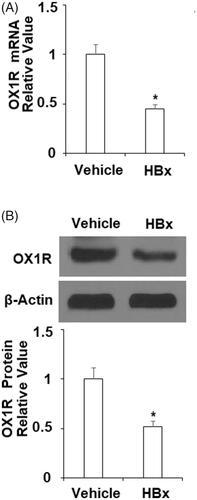
Orexin A mitigates HBx-induced oxidative stress in normal human L-02 hepatocytes
The inhibitory role of HBx protein on OX1R indicates that HBx may directly or indirectly affect the orexin signalling pathway. We investigated this pathway by employing the specific OX1R agonist orexin A in our HBx transfection experiment. HBx protein is known to induce oxidative stress in hepatic cells. Thus, we tested the possible effect of orexin A treatment on HBx-induced ROS production. 4-HNE is an indicator of ROS production and is released by lipid oxidation. As shown in and compared to control cells, HBx overexpression induced more than three-fold 4-HNE production. However, the same level of HBx induced only about 2- and 1.5-fold 4-HNE upon addition of 100 and 300 nM of orexin A, respectively. Meanwhile, as shown in , HBx overexpression resulted in cellular ROS production of about 3.5-fold. However, the same concentration of HBx resulted in only about 2.5- and 1.5-fold ROS production in the presence of orexin A, respectively.
Figure 3. Orexin A mitigates HBx-induced oxidative stress in L-02 hepatocytes. L-02 hepatocytes were transfected with the HBx-encoding plasmid for 24 h, followed by treatment with orexin A at a concentration of 100 or 300 nM for another 24 h. (A) 4-HNE expression was measured by the immunostaining; (B) ROS was measured by the DCFH-DA assay (*, #, $, p < .01 vs previous column group).
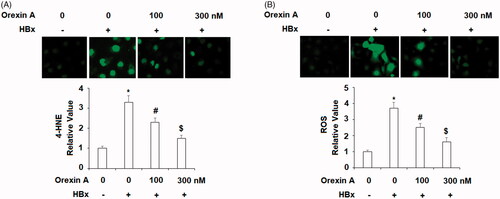
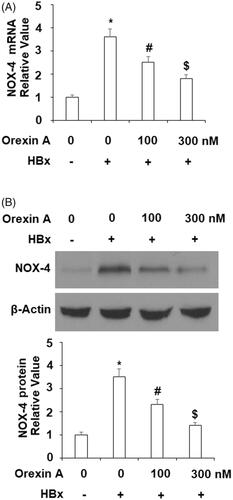
NAPDH is an important source of cellular ROS production and NOX-4 is the catalytic subunit of NAPDH oxidase. In our next orexin A experiment, we assessed changes in the level of NOX-4. Compared to the control cells, HBx expression triggered roughly 3.5-fold NOX-4 (at both the mRNA and protein levels). However, same HBx only induced about 2.5- and 1.5-fold NOX-4 in the presence of the two doses of orexin A were, respectively ( for mRNA, and for protein).
Figure 4. Orexin A ameliorates HBx-induced expression of NOX-4 in normal human L-02 hepatocytes. L-02 hepatocytes were transfected with the HBx-encoding plasmid for 24 h, followed by treatment with orexin A at a concentration of 100 or 300 nM for another 24 h. (A) Expression of NOX-4 at the mRNA levels determined by the real time analysis; (B). Expression of NOX-4 at the protein levels determined by the western blot analysis (*, #, $, p < .01 vs previous column group).
Orexin a ameliorates HBx-induced mitochondrial dysfunction in hepatocytes
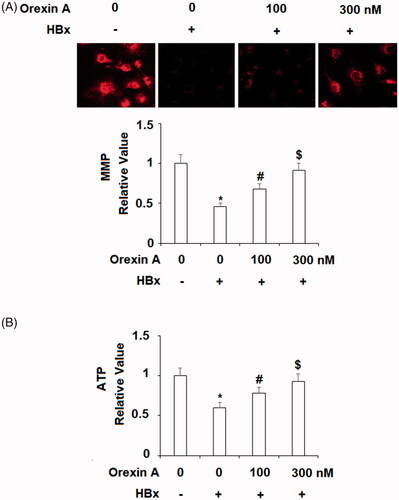
One of the mechanisms of HBV infection is that HBx targets and harms mitochondria. Next, we decided to explore the effects of orexin A on mitochondrial function. We assessed the addition of orexin A on mitochondrial membrane potential and cellular ATP production when HBx was overexpressed in L-02 cells. Compared to the control cells, HBx over-expression resulted in an approximate 55% decrease in MMP, but the two doses of orexin A recovered about 70% and 90% of the normal MMP value, respectively (). For cellular ATP production, HBx expression decreased ATP generation by about half, but two doses of orexin A recovered MMP to about 75% and 90%, respectively (). Therefore, these experiments confirm that orexin A ameliorates HBx-induced mitochondrial injury.
Figure 5. Orexin A ameliorates HBx-induced mitochondrial dysfunction in normal human L-02 hepatocytes. L-02 hepatocytes were transfected with the HBx-encoding plasmid for 24 h, followed by treatment with orexin A at a concentration of 100 or 300 nM for another 24 h. (A) Mitochondrial membrane potential (MMP) was measured by TMRM; (B) Intracellular ATP levels (*, #, $, p < .01 vs previous column group).
Orexin a inhibits HBx-induced production ofpro-inflammatory cytokines
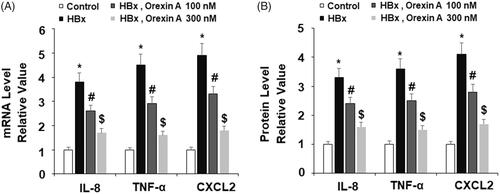
Based on the protective effect of orexin A in hepatocytes, we hypothesized that orexin A may have an anti-inflammatory role. Thus, we assessed whether orexin A could affect HBx-induced production of several key pro-inflammatory cytokines including IL-8, TNF-α and CXCL-2. Both mRNA transcripts and protein expression of these cytokines were examined in our experiments. At the mRNA level and compared to the non-treated control, HBx increased expression of these cytokines by several fold, but the addition of the two doses of orexin A significantly inhibited HBx-induced mRNA transcripts of these cytokines, and the higher dose exerted a stronger effect (). At the protein level and compared to the control, HBx still induced an increase in the expression of these cytokines of several fold, and the presence of orexin A had similar inhibitory effects on these cytokines, with the high dose exerting a stronger effect ().
Figure 6. Orexin A inhibits HBx-induced expression and secretion of pro-inflammatory cytokines in normal human L-02 hepatocytes. L-02 hepatocytes were transfected with the HBx-encoding plasmid for 24 h, followed by treatment with orexin A at a concentration of 100 or 300 nM for another 24 h. (A) Expression of IL-8, TNF-α and CXCL2 at the mRNA levels was determined by real time PCR analysis; (B) Expression of IL-8, TNF-α and CXCL2 at the protein levels was determined by ELISA assay (*, #, $, p < .01 vs previous column group).
Orexin a attenuates HBx-induced hepatocyte cytotoxicity
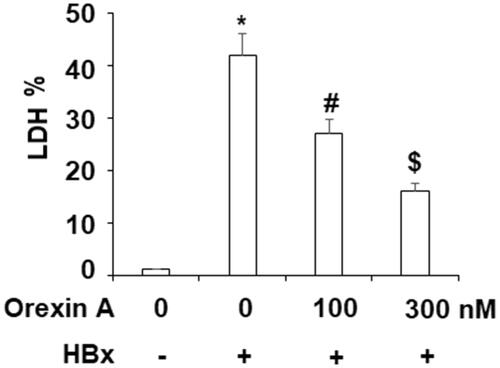
Next, we asked if orexin A would exert cellular protection against HBx-induced cytotoxicity. Indeed, our assessment of cellular LDH release revealed a protective effect of Orexin A. In our experiment, we measured free LDH levels and compared them to non-treated hepatocytes. Our results show that HBx induced more than 40% higher LDL release. However, this induction was significantly suppressed upon addition of the two doses of orexin A, increasing only about 28-fold and 15-fold, respectively ().
Figure 7. Orexin A attenuates HBx-induced cytotoxicity. L-02 hepatocytes were transfected with the HBx-encoding plasmid for 24 h, followed by treatment with orexin A at a concentration of 100 or 300 nM for another 24 h. Release of LDH was assayed (*, #, $, p < .01 vs previous column group).
Orexin a suppresses HBx-induced activation of JNK and NF-κB pathways
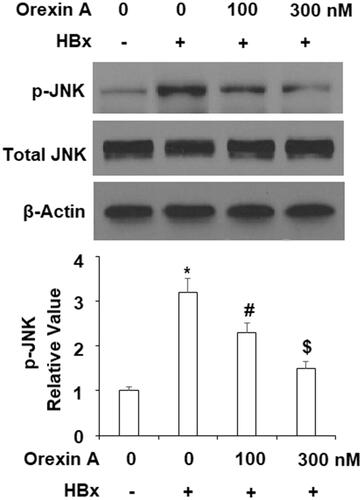
We explored the possible molecular mechanism involved in the effects of orexin A. It has been shown that HBV infection activates the JNK pathway, so we tested whether the protective role of orexin A involves the JNK pathway. In our experiment, we used the phosphorylation of JNK as an indicator of JNK activation. As shown in , when compared to the control cells, HBx induced more than 3-fold JNK phosphorylation, which was reduced to only about 2- and 1.5-fold in the presence of the two doses of orexin A, respectively, while the total JNK protein expression remained unchanged ().
Figure 8. Orexin A suppresses HBx-induced phosphorylation of JNK in L-02 hepatocytes. L-02 normal hepatocytes were transfected with the HBx-encoding plasmid. After 24 h, cells were treated with orexin A at the concentrations of 100 and 300 nM for another 2 h. Phosphorylated and total levels of JNK were determined using western blot analysis (*, #, $, p < .01 vs previous column group).
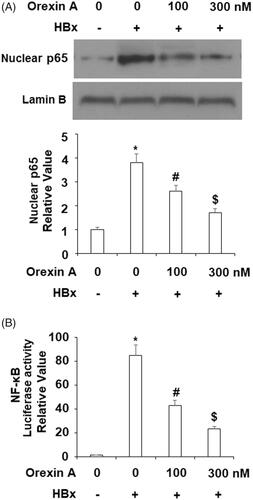
Another aspect of the mechanism of HBx is its activation of the NF-κB pathway. We tested whether orexin A could influence NF-κB activation by HBx. Two experiments were performed to address this question. We assessed the level of nuclear p65 as well as transfected NF-κB promoter activity. P65 accumulation in nuclei is the foundation of activation of NF-κB. Our experimental findings showed that the level of nuclear p65 was about 3.5-fold when HBx was overexpressed, compared to the control. However, it was only about 2.5- and 1.5-fold in the presence of the two doses of orexin A (). When we measured the activity of HBx-transfected NF-κB promoter, HBx triggered roughly 80-fold promoter activity when compared to the control. However, this induction was only about 40- and 20-fold in the presence of the two doses of orexin A (). These experiments confirmed that orexin A could suppress HBx- mediated NF-κB activation.
Figure 9. Orexin A treatment inhibited HBx-induced activation of NF-κB in L-02 hepatocytes. L-02 normal hepatocytes were transfected with HBx-encoding plasmids. After 24 h, cells were treated with orexin A at the concentrations of 100 and 300 nM for another 24 h. (A) Nuclear translocation of p65; (B) Luciferase activity of NF-κB (*, #, $, p < .01 vs previous column group).
Orexin a protects human MIHA hepatocytes against HBx-induced insults
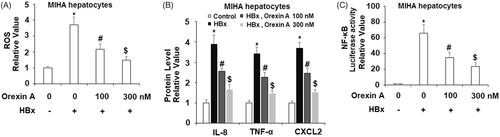
To further confirm the protective effects of orexin A against HBx-induced cytotoxicity in human hepatocytes, another type of hepatocytes, MIHA cells were used. DCFH-DA staining results indicate that treatment with 100 or 300 nM orexin A reduced HBx-induced increased ROS production in MIHA hepatocytes in a concentration-dependent manner (). Correspondingly, ELISA results demonstrate that orexin A inhibited HBx-induced secretions of IL-8, TNF-α, and CXCL2 (). Notably, the results of the luciferase activity assay in indicate that orexin A reduced HBx-induced increased NF-κB promoter activity.
Figure 10. The protective effects of orexin A against HBx-induced cytotoxicity in human MIHA hepatocytes. Human MIHA hepatocytes were transfected with the HBx-encoding plasmid for 24 h, followed by treatment with orexin A at a concentration of 100 or 300 nM for another 24 h. (A) ROS was measured by DCFH-DA assay; (B). Secretions of IL-8, TNF-α and CXCL2 were measured by ELISA analysis; (C). Luciferase activity of NF-κB (*, #, $, p < 0.01 vs previous column group).
Discussion
Orexin A is a small peptide with 33 amino acids and two disulfide bonds. Orexin A acts by activating its two GCPR receptors, OX1R and OX2R. However, OX1R has a much greater affinity for orexin A than OX2R (100–1000 times), suggesting that the two receptors play different roles [Citation7]. Indeed, the two orexin receptors play varied roles in different tissues and cells [Citation8]. Orexin A and activation of its receptors were shown to be the central regulators of food intake, energy homeostasis and wake-sleeping cycle. Recently growing evidence suggests that orexin A has a major function in peripheral tissues. According to previous studies, orexin A and its receptors are expressed in hepatocytes and involved in the regulation of normal liver function and disease progression. In rat hepatocytes, orexin A activates OX1R expression, promotes cell proliferation, and offers protection against apoptotic stimuli [Citation9]. Orexin A also stimulates glucose utilization in the liver and in cultured hepatocytes by upregulating glucose transporter GLUT-4 [Citation10]. Liver metastases have higher OX1R mRNA and the presence of orexin A induces cancer cells apoptosis [Citation11]. Orexin A also facilitates glucose flux into mitochondria and promotes oxidative metabolism in HepG2 cancer cells [Citation12]. In a prediabetes mouse model, the administration of orexin A has been shown to prevent hepatic insulin resistance [Citation13]. The most recent study showed that orexin A may contribute to the regulation of lipid metabolism in the liver as orexin A knockout mice exhibit greater weight gain [Citation14].
In our hepatocyte cell culture in vitro model, HBx expression vector was introduced into hepatocytes to mimic HBV virus infection. HBV expression is detrimental to the normal function of hepatocytes, and HBx expression causes many cellular alterations. In our study, the presence of orexin A ameliorated much of the damage resulting from HBx expression in hepatocytes. Our study began with the goal to map HBV-induced host immune response signals. Our examination of HBx expression in hepatocytes confirmed its mRNA and protein expression after plasmid transfection. We then found that OX1R expression is downregulated in hepatocytes. We confirmed that both the mRNA and protein expression of OX1R was repressed upon HBx overexpression. These facts lay the groundwork for our hypothesis that orexin A receptor-mediated signals are among the responsive factors to HBx induction. We then designed multiple experiments to rescue reduced orexin receptor signalling by treating HBx-expressing hepatocytes with orexin A and demonstrated the protective effect of orexin A against HBx expression. Our studies elucidate four facets of cellular protection by orexin A. First, orexin A ameliorates HBx-induced cellular oxidative stress. HBx is partly responsible for the 10,000-fold increase in intracellular ROS upon HBV infection [Citation15]. Our data show that orexin A suppresses HBx-induced 4-HNE release, NOX-4 expression and total cellular ROS production. 4-HNE is one of the major products of lipid peroxidation and has been shown to be present at different stages of pre-carcinogenic hepatitis [Citation16]. NOX-4 is the dominant catalytic unit of NAPDH oxidase in hepatocytes, which attributes to liver fibrosis and hepatic diseases [Citation17]. These facts demonstrate that orexin A possesses anti-ROS property and relieves HBx-induced oxidative stress. Thus, there may be a potential liver protection mechanism mediated by orexin A. Second, our data show that orexin A protects against HBx-induced mitochondrial dysfunction as revealed by its amelioration of the collapse of mitochondrial membrane potential and reduced cellular ATP. The liver is the chemical centre of the human body and hepatocytes are enriched with mitochondria that comprise about 13–20% of the liver volume [Citation18]. HBx directly targets mitochondria and alters its function [Citation19]. The capability of orexin A to ameliorate HBx-induced mitochondrial dysfunction suggests that mitochondrial rescue is one of mechanisms behind orexin A-mediated hepatocyte protection. Third, our data show that orexin A inhibits the release of major pro-inflammatory cytokines including IL-8, TNF-α, and CXCL2. The inhibitory effect of orexin A on these inflammatory factors suggests that it may be able to suppress HBV-induced hepatocyte inflammation. Fourth, orexin A relieves HBx-induced cellular cytotoxicity as revealed by reduced LDH release. The amelioration of orexin A on HBx-induced cytotoxicity may contribute to its anti-ROS, anti-inflammation and mitochondrial protection in hepatocytes. Mechanistically, orexin A and its receptors transmit cellular signals via the JNK and NF-κB pathways. In liver diseases including hepatitis and hepatocellular carcinoma, the interaction mechanism of the JNK and nuclear NF-kB pathways is critical for disease development [Citation20]. The capability of orexin A to inhibit activation of these two pathways indicates its potential beneficial effects against HBV infection and hepatitis.
In conclusion, orexin A exhibits robust modulation of hepatocyte function and could have certain beneficial effects in liver diseases. Our study explicitly shows that orexin A activates its receptor signals and protects against harm from HBV infection. An exciting prospect for our next experiment would be to establish an HBV animal model to evaluate its effect in vivo.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- MacLachlan JH, Cowie BC. Hepatitis B virus epidemiology. Cold Spring Harb Perspect Med. 2015;5:a021410
- Liang TJ. Hepatitis B: the virus and disease. Hepatology. 2009;49:S13–S21.
- Terrault NA, Bzowej NH, Chang KM, et al. AASLD guidelines for treatment of chronic hepatitis B. Hepatology 2016;63:261–283.
- Seeger C, Mason WS. Molecular biology of hepatitis B virus infection. Virology. 2015;479–480:672–686.
- Carrive P. Orexin, orexin receptor antagonists and central cardiovascular control. Front Neurosci. 2013;7:257.
- Xu TR, Yang Y, Ward R, et al. Orexin receptors: multi-functional therapeutic targets for sleeping disorders, eating disorders, drug addiction, cancers and other physiological disorders. Cell Signal. 2013;25:2413–2423.
- Ebrahim IO, Howard RS, Kopelman MD, et al. The hypocretin/orexin system. J Royal Soc Med. 2002;95:227–230.
- Leonard CS, Kukkonen JP. Orexin/hypocretin receptor signalling: a functional perspective. Br J Pharmacol. 2014;171:294–313.
- Ju SJ, Zhao Y, Chang X, et al. Orexin A protects cells from apoptosis by regulating FoxO1 and mTORC1 through the OX1R/PI3K/AKT signaling pathway in hepatocytes. Int J Mol Med. 2014;34:153–159.
- Zhang C, Sun C, Wang B, et al. Orexin-A stimulates the expression of GLUT4 in a glucose dependent manner in the liver of orange-spotted grouper (Epinephelus coioides). Comp Biochem Physiol A Mol Integr Physiol. 2016;199:95–104.
- Voisin TE, Firar A, Fasseu M, Rouyer-Fessard C, et al. Aberrant expression of OX1 receptors for orexins in colon cancers and liver metastases: an openable gate to apoptosis. Cancer Res. 2011;71:3341–3351.
- Liu Y, Zhao Y, Guo L. Effects of orexin A on glucose metabolism in human hepatocellular carcinoma in vitro via PI3K/Akt/mTOR-dependent and -independent mechanism. Mol Cell Endocrinol. 2016;420:208–216.
- Tsuneki H, Tokai E, Nakamura Y, et al. Hypothalamic orexin prevents hepatic insulin resistance via daily bidirectional regulation of autonomic nervous system in mice. Diabetes. 2015;64:459–470.
- Mochizuki A, Nakayama K, Nakamura S, et al. Involvement of orexin in lipid accumulation in the liver. J Oral Biosci. 2018;60:76–82.
- Ivanov AV, Valuev-Elliston VT, Tyurina DA, et al. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget. 2017;8:3895–3932.
- Marquez-Quiñones A, Cipak A, Zarkovic K, et al. HNE-protein adducts formation in different pre-carcinogenic stages of hepatitis in LEC rats. Free Radic Res. 2010;44:119–127.
- Liang S, Kisseleva T, Brenner DA. The role of NADPH oxidases (NOXs) in liver fibrosis and the activation of myofibroblasts. Front Physiol. 2016;27:17
- Berry MN, Edwards AM. The hepatocyte review. Dordrecht: Kulwer Academic Publishers; 2000.
- Lim W, Kwon SH, Cho H, et al. HBx targeting to mitochondria and ROS generation are necessary but insufficient for HBV-induced cyclooxygenase-2 expression. J Mol Med. 2010;88:359–369.
- Papa S, Bubici C, Zazzeroni F, et al. Mechanisms of liver disease: cross-talk between the NF-kappaB and JNK pathways. Biol Chem. 2009;390:965–976.