
Abstract
Atherosclerosis is a complex disease with involvement of intermediate-, large-sized arteries. Atherosclerosis is characterized by the accumulation of vascular lipids, immune system activation, inflammation, oxidative stress and oxidized low-density lipoproteins (LDLs), endothelial cell (EC) activation, arterial smooth muscle cell (SMC) proliferation, macrophage activation and foam cell formation that cause endothelial dysfunction. DNA methylation is one of important epigenetic mechanisms which changes gene expression. It has been evident that this mechanism plays an important role in the initiation and propagation of atherosclerosis. Furthermore, DNA methylation is a crucial and distinct mechanism that modulates genes governing cell proliferation, thereby connecting environmental insults with gene expression. This study represents many atherosclerosis-related genes which are regulated through DNA methylation mechanism. Although the role of epigenetics in atherosclerosis is at their infancy. Nevertheless, various studies demonstrated that DNA methylation involvement in this disease is undeniable. DNA methyltransferases are the main player of the smooth muscle cell proliferation, endothelial cell integrity, as well as arteriosclerosis formation. In this review, we focus on recent achievements in the functional and description interpretation of the DNA methylation pattern of cells and tissues implicated in atherosclerosis. Besides, we discuss the association of DNA methylation with oxidative stress, hyperhomocysteinemia (HHcy), ageing, and inflammation in the development and pathogenesis of atherosclerosis.
Introduction
Atherosclerosis is the main cause of many cardiovascular diseases, including ischemic gangrene, coronary artery disease (CAD), abdominal aortic aneurysms, and many cases of stroke and heart failure [Citation1]. Atherosclerosis-related diseases still are the main cause of morbidity and mortality in the Western world and also has been the primary cause of death for the past 50 years [Citation2]. Studies show that more than 40% of deaths in the United States are due to cardiovascular disease [Citation3]. Atherosclerosis is a slow and dynamic process which cellular and molecular activities are implicated within the atherosclerotic plaques [Citation4]. Atherosclerosis is a complicated phenomenon with a complex process which starts with accumulation of vascular lipids, immune system activation, inflammation with inflammatory cytokine such as TNFα and IL-1, oxidative stress and the infiltration of oxidized low-density lipoproteins (LDLs), as well as endothelial cell (EC) activation, proliferation of arterial smooth muscle cell (SMC), macrophage activation and foam cell formation that cause endothelial dysfunction [Citation5–10].
The main and first strategies for atherosclerosis treatment are mainly medications to lower cholesterol and decrease clotting and procedures such as percutaneous coronary intervention, carotid endarterectomy or coronary artery bypass graft. Due to the effectiveness of the treatments, there are still patients with atherosclerosis-related diseases who still suffer extreme pain in their chest and also their quality of life is not good enough [Citation11]. Besides the progresses which are made in this regard, there is still no comprehensive treatment for this disease, and further research is needed to figure out a new and comprehensive treatment for these patients.
DNA methylation is one of important epigenetic mechanisms which causes gene suppression [Citation11]. DNA methylation involves in many cellular phenomena such as X-chromosome inactivation, transposable retro element activities, cell differentiation, cell reprogramming, cell death, cell survival, parental gene imprinting, immune system activities and allelic exclusion of immunoglobulin genes [Citation12,Citation13]. DNA methylation abnormalities are linked with different diseases such as cancer, autoimmune diseases and atherosclerosis [Citation14–19]. Investigations on DNA methylation in patients with atherosclerosis documented that there is a unique profile of DNA methylation in this disease, and emphasize on the different pathways and genes which are implicated in the disease [Citation20]. More researches are required to illustrate the DNA methylation signature and the underling mechanism of these modifications in atherosclerosis disease. With clarification and characterization of these abnormalities, we hope that this knowledge may be applied in the diagnosis and treatment of atherosclerosis patients. This review emphasizes on the recent knowledge in DNA methylation abnormalities and their roles in the pathogenesis and progression of atherosclerosis.
DNA methylation
DNA methylation is a process which a methyl group is added to the carbon of cytosine at 5’ position in a CpG site. Actually, methyl group is added to a cytosine-paired-with-guanine (CpG) dinucleotide sequences through DNA methylation process [Citation21]. Furthermore, methyl group could have added to a cytosine residue which is not located adjacently to a guanine residue [Citation22]. Most of CpG sites (more than 70%) are methylated in the genome, which are actually distributed throughout the majority of the genome including transposable elements, endogenous repeats and gene bodies. CpG islands, dense and high number of CpG sites in a short area of DNA, are mainly located at the promoter region and generally are unmethylated. The main characteristics of a CpG island are; at least 200 bp in length, GC content more than 50%, and the observed/expected ratio of CpG frequency more than 0.6 [Citation23].
DNA methyltransferase (DNMT) is a family of enzyme which is responsible for DNA methylation and traditionally divided into two subgroups; de novo methylation and maintenance methylation. De novo DNMTs consist of DNMT3a and DNMT3b and the only maintenance DNMT is the DNMT1. DNMT3a and DNMT3b are upregulated in embryonic stem cells and have a low expression in differentiated cells. During the early embryonic development and following embryo implantation, DNMT3a and DNMT3b are responsible to add a methyl group to DNA to previously unmethylated DNA. The methyl group is provided by S-adenosylmethionine (SAM), the universal methyl donor. To the establishment of methylation patterns in the early development embryonic stage, de novo methylation is critical. Through a cell division, the methylation signature of the original and old stand of DNA is copied into the new strand of DNA by DNMT1, which is the main DNMT in the cell. This process is named as methylation maintenance. DNMTs through interaction with epigenetic factors or regulators and recognizing specific chromatin structures targets a specific site within the genome and methylated the particular region of genome [Citation24].
DNA methylation through modulating the interaction between DNA and proteins regulates gene transcription. DNA hypermethylation usually leads to a long-term and stable gene suppression until DNA hypomethylation of the gene leads to gene expression. DNA methylation modulates gene expression through two different approaches. The first described way is that the methylated CpG sites impede the attachment of transcription factors to the DNA. The next way is that a family of methyl-CpG binding domain (MBD) proteins, Kaiso (nonhomologous protein) and four homologous proteins (MeCP2, MBD1, MBD2, and MBD4), can bind to the methylated CpG sites.
The MBD proteins through interaction with other proteins and heterochromatin formation and also by direct prevention of transcription factor to bind to the promoter region leads to gene silencing.
These MBD proteins recruit enzymes such as HDAC (histone deacetyl transferase) that catalyze histone deacetylation and heterochromatin formation. In contrast, hypomethylation of genome generates a euchromatin formation which is favourable for gene transcription by interacting with histone acetylation [Citation25,Citation26].
Aberrant DNA methylation and diseases
Studies showed that hypermethylation of DNA is implicated in various diseases. In the healthy individuals, CpG islands in the promoter region of genes are usually hypomethylated, whereas the CpG islands in non-promoter region are generally hypermethylated. Global DNA hypomethylation which is known as DNA hypomethylation of non-promoter regions may cause structural changes and instability of chromosomes because of the initiation of transcription at incorrect region and high transcriptional activity in normally silent sites. The global DNA hypomethylation leads to expression of potentially harmful genes and also, high expression of normally silenced genes. At the other hands, global DNA hypermethylation causes inactivation of disease-suppressor-gene or protective-gene, genes mutation and allelic loss. For instance, low expression of transposable elements, such as short interspersed nucleotide element (ALU) and long interspersed nucleotide elements-1 (LINE-1) in non-promoter regions, are documented to play pivotal roles in the maintenance of genome integrity through their heavy methylation. Methylation of these transposable elements could be considered as a surrogate marker of DNA methylation signature of genome [Citation24]. In cancers, these transposable elements are highly hypomethylated and this change causes chromosomal instability, DNA recombination, insertional mutagenesis, and these changes might be implicated in cancer pathology [Citation27]. Hypermethylation of DNA in the promoter region of tumour suppressor genes is a well-studied epigenetic marker in various cancers [Citation28]. Since there are many similarities between cancers and CVDs such atherosclerosis, it is thought that the dysregulation of cardiovascular gene expression might also be implicated in atherosclerosis.
Oxidative stress and DNA methylation in atherosclerosis
We are exposed to different exogenous and endogenous reactive oxygen species (ROS) in our life. The imbalance between daily ROS generation and antioxidant systems leads to oxidative stress. In a healthy situation, the balance between ROS formation and enzymatic and non-enzymatic antioxidant elements are maintained and the ROS are reduced or scavenged through our antioxidant system [Citation29]. The impairment in the antioxidant system or an elevated ROS production could lead to a redox imbalance [Citation30]. Chronic oxidative stress is an underlying cause of ageing and diseases such as cancers, cardiovascular disorders, inflammation, and infection [Citation31–33]. Various researches have documented that oxidative stress through atherosclerosis progresses could modulate DNA methylation [Citation34–36]. The idea that oxidative stress might modulate DNA methylation came from the facts that the cancer cells are often in an oxidative stress environment and also, their methylation signature is totally changed [Citation37]. There are studies which showed that the presence of ROS could affect the methylation pattern. For example, in 1994, an in vitro experiment showed that the 8-hydroxyguanine (8-OHdG), indicator of ROS, could negatively change the methylation status of DNA [Citation38]. Another study suggests that oxidative damage of guanines by 8-OHdG in the parental strand would permit normal copying of methylation patterns through DNA methylation maintenance mechanism, while oxidative damage of guanines by 8-OHdG in the new DNA strand would impede such methylation [Citation39]. Generally, these investigations propose that damage from oxidative stress on the new DNA strand could impede DNA methylation. 5-hydroxymethylcytosine (5hmC) is a molecule which inhibits DNA methylation and leads to DNA hypomethylation [Citation40]. O'Hagan HM and their colleagues showed that ROS especially hydrogen peroxide can change the methylation pattern of DNA [Citation41]. They reported that H2O2 treatment, through binding of DNA methyltransferase1 (DNMT1) to chromatin, can change the methylation status of CpG sites. In case of atherosclerotic lesions, DNA methylation signature could be changed by ROS in a similar way. In addition, superoxide dismutase 2 (SOD2) gene suppression through DNA methylation mechanism causes proliferation of SMC. Treatment with the 5-azacytidine (5-AZA), the DNMT inhibitor, leads to high expression of SOD2 and reduced proliferation of SMC through DNMT activity. Since methylation of SOD2 could activate HIF-1α, its methylation is critical in the development of atherosclerosis plaques and an apoptosis-resistant and proliferative microenvironment [Citation42].
ROS through change in DNMT activity and DNA damage causes changes in DNA methylation. ROS compounds are extremely reactive and oxidize macromolecules such as DNA. Many different damaged sites will be generated through DNA oxidation. One of the most commonly generated base lesion damage is 8-OHdG, which is applied as a marker of oxidative damage in DNA [Citation43]. Modification of DNA methylation in nearby cytosine bases and an elevated mutation are two main consequence of oxidative damage of DNA. A study by Valinluck and their colleagues showed that hydroxylation of a cytosine causes 90% decrease in its methylation [Citation40]. These observations have shown that the amount of methylation during the cell division will be reduced and ultimately leads to the formation of a methylome, which is apparently characterized by global hypomethylation. 5hmC formation from 5-methylcytosine (5mC) through intracellular enzymes has been evident over the past years [Citation44]. Although, it has not been clear yet, whether all these hydroxymethylated CpG sites are result of incorrect methylation signatures. Although DNA methylation is a protected mechanism, oxidative damage can disrupt this carefully maintained mechanism [Citation45]. In addition, hydroxylated 5mC may be incorrectly converted to unmethylated cytosine via DNA repair mechanisms, which is thought to be one of the mechanisms that actively demethylated cytosine in the non-dividing cells [Citation13]. It has not been provided the large-scale analytical techniques to evaluate the genome-wide hydroxymethylation signature. Therefore, there is not much detail yet about this process and its frequency. Methods that applied to evaluate the whole genome methylation signature cannot distinguish the methylated from hydroxymethylated cytosine [Citation46]. Although 5hmC has been shown to be different from 5mC in terms of its function and interaction with methyl-CpG binding proteins, the effect of ROS-induced hydroxymethylation on the epigenome is still unclear [Citation47]. ROS have various impacts on methylation such as; change the expression of DNMTs, induces the differential binding of DNMT-containing complexes and catalyzes the DNA methylation.
On the one hand, Vilkaitis and colleagues suggested a mechanism by which they showed that ROS thereby lead to hypermethylation of DNA in the promoter regions [Citation48]. The fact is that the hypermethylation of promoter regions is comparable to other genome regions, which can explain how this specific, stochastic mechanism leads to hypermethylation in a particular position. Nonetheless, carefully controlled investigations are required to evaluate the ROS ability to induce the cytosine methylation in the absence of DNMTs and presence of S-adenosylmethionine (SAM).
On the other hand, O'Hagan et al. illustrated that treatment of cancer cells with H2O2 leads to CpG island-containing low-expression genes gain promoter DNA methylation through the formation of DNMT1 and DNMT3b. They suggest that oxidative damage induces relocalization and formation a silencing complex which causes DNA hypermethylation. Furthermore, they proposed an explanation for the higher binding capacity of the silencing complex to CpG islands than to non-CpG islands and this mechanism may explain how tumour suppressor genes are hypermethylated and silenced [Citation41]. Furthermore, Griffiths et al. showed that in the oxidative stress situation, DNA methylation and expression of DNMT1 and DNMT3b were elevated in the transformation of a melanocyte cell line [Citation49]. Investigations about the effect of oxidative stress on methylation of DNA have been largely analyzed by different researchers. According to our best knowledge, chronic oxidative stress may modify the methylation signature of DNA through the pathological development of the disease. It has been proven that there are mechanisms which oxidative stress through them modifies DNA methylation. Since the methylation pattern is changed through oxidative stress, our present article documented that oxidative stress is an important risk factor for atherosclerosis pathogenesis.
DNA methylation and inflammation in atherosclerosis
Atherosclerosis is an inflammation-based disorder which is characterized by the adherence of blood circulating cells such as monocytes to the endothelium, then migrate to the sub-endothelial layer, and differentiate into the monocyte-derived macrophages. Low-density lipoprotein (LDL) particles in blood plasma invade the endothelium and become oxidized through a set of biochemical reactions, creating risk of atherosclerosis. Initial damage to the endothelium causes an inflammatory response. Immune cell migration is mediated through selectin and integrin interaction with their ligands, M-CSF and other cytokine in the site of injury leads to differentiation of macrophages from monocytes. Activated macrophages turn into the “foam cells” after their local proliferation and digestion of oxidized LDL. Foam cells eventually die and exacerbate the inflammatory condition [Citation50].
Investigations in clinic and in vitro studies reported that even earliest lesions of atherosclerosis are associated with leukocyte infiltration. Vasoactive molecules, such as endothelins, eicosanoids and nitric oxide are released from activated macrophages in the atheroma [Citation51,Citation52]. Furthermore, ROS, pivotal cause of cytotoxicity and lipoprotein oxidation, is another molecule which is released from the activated macrophages [Citation53]. Ox-LDL particles, an independent risk factor for atherosclerosis, propagate inflammation through activation of vascular ECs [Citation53,Citation54]. Various studies documented that inflammatory mediators in atheroma such as cyclooxygenase-2 (Cox-2), CD40 ligand, tumour necrosis factor α (TNF-α) and interleukin-1b can increase expression of matrix metalloproteinase in smooth muscle, endothelial and phagocyte cells. Many studies are applied to evaluate the role of epigenetic mechanisms in gene transcription. Nevertheless, there is not enough research in case of epigenetic involvement in the regulation of inflammatory and anti-inflammatory genes.
DNA methylation, an important epigenetic mechanism, modulates the expression of genes which are connected to atherosclerosis and inflammation. Alteration in DNA methylation has been associated with chronic inflammatory profile of diseases. An investigation documented that inflammatory condition is associated with hypermethylation of DNA; and also, further analysis showed that DNA hypermethylation is connected to an increased mortality rate in atherosclerosis-related diseases [Citation55]. Hypomethylation of Toll-like receptor 2 (TLR-2) promoter region is linked to an elevated inflammatory immune response [Citation56]. Luminometric technology was applied by Stenvinkel et al. to evaluate the global genomic DNA methylation [Citation57]. They showed that there is a strong correlation between DNA methylation and inflammation. Another example of the relationship between cardiovascular disease-related inflammation and DNA methylation is the expression of cytochrome C oxidase subunit II (Cox-2) gene. Cox-2 associated with the development of atherosclerosis and its expression can be induced by proinflammatory cytokine such as TNF-α. An investigation by Tong et al. [Citation58] documented that there is a negative correlation between protein synthesis and mRNA expression of Cox-2 with DNA methylation. They showed that epigenetic components have an important role in the regulation of Cox-2 expression.
Studies reported that a low level of COX-2 expression in high-risk aspirin-treated patients may protect against atherothrombosis [Citation59]. The prostanoid biology has multifaceted aspects; and also, COX-2-derived PGI2 has an important role in systemic hemodynamics in the setting of inadequate circulatory volume. These issues should be considered when evaluating the benefits of COX-2 inhibition. Furthermore, the downstream and upstream regulatory sites of COX-2 expression should be considered.
Moreover, it has been reported that NF-κB-mediated signalling is regulated through DNA methylation. Inflammation in cardiovascular diseases can change DNA methylation and is a susceptible risk factor for atherosclerosis development [Citation54]. On the other hand, methylation can also cause inflammation through DNMT, which might accelerate the atherosclerosis progression.
DNA methylation and ageing in atherosclerosis
Atherosclerosis and ageing are two important major independent susceptible risk factors for mortality and morbidity of cardiovascular diseases. Studies showed that clinical manifestations of atherosclerosis are connected to ageing process [Citation60,Citation61]. Autopsy results from vascular beds supplying blood to the kidneys, heart, lower extremities and brain demonstrated that atherosclerosis and aging are closely related [Citation62]. Although it has been shown that there are many changes in gene expression with ageing process, but the cause of these changes is not clear yet. Evaluation of DNA methylation as an important epigenetic mechanism has led to speculation that it might be implicated in the ageing process [Citation63]. Studies showed that in vitro passage of normal fibroblast led to DNA hypomethylation [Citation64,Citation65]; and also, in vivo aged tissues are similarly hypomethylated [Citation66–68]. These investigations reported that ageing process could lead to hypomethylation of genome. On the other hands, hypermethylation at specific sites has been reported such as E-cadherin, c-Fos, P15, ERα, PAX6, c-Myc, DBCCR1, insulin-like growth factor-II, versican, MYOD1, HIC1, and N33 [Citation69]. Interestingly, many of genes which had been previously thought to be methylated only in cancer have now been found to belong to the group of age-related methylated genes. In fact, investigations illustrated that the common cause of hypermethylation in cancer is actually the age-related methylation [Citation69]. For example, in case of ERα methylation in heart muscle the underlying cause is age-related methylation [Citation70]. In cultured SMCs, the methylation status of ERα in the first passage in a sample from an infant and an adult cadaver was totally different range from 19% to 99%, respectively [Citation71]. Therefore, age-related methylation is not an exclusive feature of cancer; rather, it may involve in other age-related diseases, such as atherosclerosis. In addition to age-related global hypomethylation, gene-specific hypermethylation has been reported and is associated with the instability of genome and high rate of mutation. Evaluation of DNA methylation of aortas in atherosclerotic and normal arteries of rabbit showed that DNA is hypomethylated in atherosclerotic aortas in comparison to normal arteries [Citation72]. Thus, it concludes that age-related global hypomethylation may have an important effect on the atherosclerosis pathogenesis.
DNA methylation and SMCs in atherosclerosis
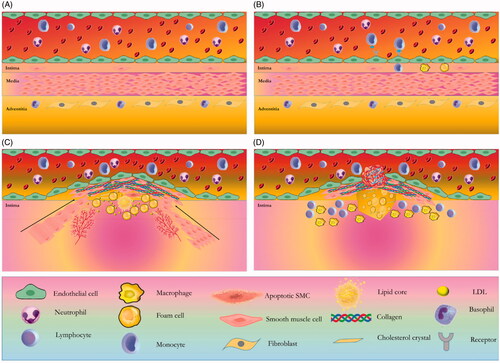
Pathologic process of atherosclerosis consists of three important stages (). The first stage is EC (endothelial cell) activation through metabolic susceptible risk factors such as hyperlipidemia. ECs activation leads to infiltration of immune cells such as monocytes into the intima. Phagocytosis of LDL cholesterol causes the formation of foam cells and phospholipid oxidation. In the second stage, inflammatory condition and through the effect of mediators resulted in the infiltration of SMCs into the intima and its proliferation leads to plaque formation. In other word, theses activated SMCs produces lots of extracellular matrix and form a fibrous cap [Citation73]. Increased level of oxidized phospholipids leads to proliferation and apoptosis of vascular SMC (VSMC) [Citation74–77]. The third and final stage is atherosclerotic plaque rupture and thrombosis. Since the VSMCs are the main player of atherosclerotic plaque formation, regulation of VSMC could play an important role in atherosclerosis management. Early development of atherosclerotic lesion is mediated through differentiation and proliferation of VSMCs and these plaques are maintained by the fibrous cap [Citation77]. Matrix metalloproteinases (MMPs) are connected to VSMC growth and as well as, atherosclerosis. For example, it has been documented that the MMP-9 is implicated in the migration of VSMCs [Citation78–80]. Although MMP-9 leads to primary lesions in atherosclerosis; on the other hand, it prevents progression of the complete lesion [Citation81,Citation82], illustrated that these VSMCs participate in increase of cholesterol influx, decrease of efflux and formation of foam cells in early stage of atherosclerotic lesions. In addition of VSMC function, their senescence and apoptosis are implicated in the atherosclerosis development. These processes are connected to coagulation, calcification, plaque rupture and vessel remodelling. In the bottom line, SMCs are evidently implicated in the formation of atherosclerotic plaque by their migration, proliferation, matrix synthesis, foam cell formation, senescence and apoptosis. Furthermore, there are many changes in protein synthesis and gene expression of SMCs. Assessment of gene expression signature of SMCs illustrated that a set of genes are transcriptionally controlled through epigenetic mechanisms. Epigenetic mechanisms could regulate gene expression profile of SMCs [Citation83,Citation84] and various researches documented that some of SMC genes are controlled through DNA methylation. Interestingly, these genes could be linked with migration, differentiation, phenotypic switching of SMCs and also the progression of vascular disorders. Some of these genes are involved in differentiation of SMC and regulated through DNA methylation include the serum response factor (SRF), platelet-derived growth factor (PDGF), ERα, ERβ, and SMC-specific SM22α, [Citation85]. An assessment on proliferating intimal SMCs of New Zealand rabbits reported a global DNA hypomethylation [Citation72]. Another study on ApoE−/− mice lesions and atherosclerotic lesions of human showed a hypomethylation profile of genome [Citation86]. It has been reported that low activity of DNMT and global DNA hypomethylation occurs during proliferation and phenotypic switch of SMC in the culture media [Citation87,Citation88]. While DNA methylation through extracellular matrix affects the SMC phenotype, phenotypic switch of SMC causes vascular calcification [Citation89].
Figure 1. Four stages in the atherosclerotic lesion formation. (A) In the normal and healthy situation, the normal artery has three layers. The intima layer or the inner layer consists of a monolayer of endothelial cells and resident smooth muscle cells (SMCs). The tunica media or the middle layer has several layers of SMCs which are embedded in a complex extracellular matrix. The adventitia, the outer layer, contains microvessels, nerve endings, mast cells, and fibroblasts. (B) The first stage is ECs activation through metabolic susceptible risk factors such as hyperlipidemia, adhesion of blood leukocytes such as monocytes to the activated monolayer of ECs, migration of monocyte into the intima, maturation of monocytes into macrophages, phagocytosis of LDL cholesterol that causes the formation of foam cells and phospholipid oxidation. (C) In the second stage, inflammatory condition and mediator effect leads to infiltration of SMCs from the media to the intima. High proliferation of media-derived SMCs and resident intimal SMCs, and an elevated production of extracellular matrix macromolecules, such as proteoglycans, elastin, and collagen leads to plaque formation. SMCs and plaque macrophages may die in advancing lesions through apoptosis. Extracellular lipid derived and apoptotic bodies from dying and dead cells can exacerbate the inflammatory condition. Advancing plaques also contain microvessels and cholesterol crystals. (D) The third and final stage is atherosclerotic plaque rupture and thrombosis, where it can impede blood flow.
Since the proteins of the ten-eleven-translocation (TET) family are implicated in demethylation of DNA [Citation44], evaluation of the role of these proteins in the phenotypic change of SMC is mandatory. TET proteins (TET1–TET3) are kind of DNA demethylases that oxidize 5mC and form 5hmC. Evaluation on 5-AZA, chemical analogue of cytidine and inhibitor of DNA methylation, showed that expression and protein secretion of MMP1 are elevated after treatment of cultured human vascular SMCs with 5-AZA [Citation90]. Furthermore, the investigation showed that 5-AZA inhibits the proliferation and migration of airway SMC which is induced by PDGF and also, increase the cellular contractility [Citation91]. These investigations documented that DNA methylation of SMC plays an important role in atherosclerosis development. Generally, hypomethylation of DNA in SMC leads to migration and proliferation of these cells and finally worsen the plaque formation.
DNA methylation and hyperhomocysteinemia (HHcy) in atherosclerosis
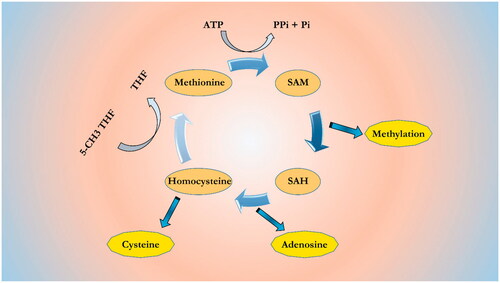
Homocysteine (Hcy), homologue of cysteine, is not a classic amino acid and derived from the methionine–homocysteine cycle [Citation92] (). In this cycle, methionine converts to S-adenosylmethionine (SAM) through enzymatic process. SAM is the main methyl donor for more than 50 different methyltransferases. The SAM molecules convert to S-adenosylhomocysteine (SAH) by a demethylation process and become a powerful competitive methyltransferase inhibitor. Enzymatic hydrolysis of SAH, form an Hcy and an adenosine. Hcy through the methylation process (5-methyltetrahyfolate (5-CH3THF)) can form methionine or also, forms cysteine through transsulfuration [Citation93]. The normal range of Hcy in plasma is 5–15 μM in healthy person [Citation94]. High concentration of Hcy in plasma is named hyperhomocysteinemia (HHcy), which categorized into three levels: when the Hcy concentration range 15–30 μM is called a moderate Hhcy, concentration range 31–100 μM is called intermediate Hhcy, and concentration more than 100 μM is called severe Hhcy [Citation95]. Hcy has been involved as an independent and common susceptible risk factor for atherosclerosis development and is implicated in VSMC proliferation and endothelial dysfunction through DNA methylation. DNA methylation abnormality is reported in patients with cardiovascular diseases concomitant with high level of Hcy [Citation96]. It has been shown that Hhcy through DNA methylation causes change in the expression of genes which is linked to atherosclerosis development. A study showed that treatment of VSMCs with high concentration of Hcy leads to hypomethylation of Alu and LINE-1 elements [Citation97]. This study reported that high concentration of Hcy may increase the DNMT activity and also, increases SAH and decreases SAM concentration. Another study reported that treatment of VSMCs with different concentrations of Hcy leads to hypomethylation and high expression of PDGF by a dose-dependent manner, thereby high proliferation rate of VSMC [Citation98,Citation99]. Hcy also, affects the estrogen receptor (ESR), an atherosclerosis-related gene. A study by Huang et al. [Citation100] analyzed the relationship among markers such as Hcy concentration, methylation of ESR1, and the severity of plaque lesions. Data analysis documented that there are positive correlations between Hcy levels with prompter methylation of ESR1 and severity of lesions. High concentration of Hcy leads to atherosclerosis initiation by oxidative stress, activation of immune system and proliferation of VSMC [Citation101,Citation102].
Figure 2. Methionine-homocysteine cycle. SAM is synthesized from ATP and methionine. SAM is the common methyl donor for almost 50 different methyltransferases that recognize different methyl acceptors, such as proteins and nucleic acids. The SAH, demethylated product of SAM, is a powerful competitive methyltransferase inhibitor. Its accumulation is prevented by SAH hydrolase to homocysteine and adenosine. Homocysteine can be remethylated to methionine by 5-methyltetrahyfolate (5-CH3THF) or trans-sulfurated to cysteine.
A study by Yideng et al reported that treatment of monocyte with Hcy causes DNA methylation of PPAR α, γ promoter region and eventually, low expression of mRNA and protein levels. The peak effect of Hcy was in the 100 μM, but, the effect was not dosed dependent. In addition, the SAM level was decreased and the SAH level was increased. These results propose that DNA methylation of PPAR α, γ, which is induced by Hcy may represent a critical mechanism to atherosclerosis development and could be a therapeutic target for preventing Hcy-induced atherosclerosis [Citation103]. Another study by Jamaluddin et al. showed that HHcy causes DNA hypomethylation of atherosclerosis-related genes such as cyclin A, which blocks cell cycle progression and regeneration of endothelium. Furthermore, they showed that the DNMT1 activity is decreased and the high expression of DNMT1 reverses the preventive effect of Hcy on cyclin A expression and EC growth inhibition. They suggested that DNA hypomethylation of cyclin A, induced by Hhcy, is an important underlying mechanism responsible for EC growth inhibition and contributes to atherosclerosis [Citation104]. Study by Yi et al. [Citation105] documented that clinically relevant Hcy levels (100 mM) may elevate cholesteryl ester (CE), free cholesterol (FC) total cholesterol (TC) concentrations in plasma. Treatment of primary human monocytes with this level of Hcy downregulates the expression and protein level of ApoE. These results proposed that DNA hypermethylation of ApoE which is instigated by HHcy may play an important role in ApoE expression in atherosclerosis [Citation106]. Abnormal lipid deposition in the proximal aorta induced by Hhcy is a sign of atherosclerosis progress. Hcy can also affect the methylation profile of genes which are contributing in cholesterol efflux. High concentration of Hcy may elevate global hypomethylation of DNA, which regulates the expression pattern of atherosclerosis-related genes. However, the mechanism by which Hcy modulates DNA methylation and the relationship between gene-specific and global DNA methylation remains unclear. Therefore, further investigations are required to determine the underlying mechanisms of gene-specific methylation, which is induced in the presence of Hcy. Future studies in this field may determine the novel therapeutic targets to prevent Hcy-induced atherosclerosis.
Conclusions
Atherosclerosis is a complex chronic disease that genetic and non-genetic factors are involved in its aetiology. One of the important non-genetic factors is DNA methylation, which is the most important epigenetic mechanism. It has been reported that DNA methylation is implicated in atherosclerosis development. Various studies reported that DNA methylation is connected to the pathogenesis of atherosclerosis. This study represents many atherosclerosis-related genes which are regulated with DNA methylation mechanism (). Besides, other epigenetic mechanisms such as non-coding RNAs and histone modifications have an important role in the atherosclerosis development and these epigenetic mechanisms may be applying as a novel and promising target for atherosclerosis treatment in the near future. Although, the role of epigenetics in atherosclerosis is at their infancy. Nevertheless, various studies demonstrated that DNA methylation involvement in this disease is undeniable. Furthermore, the application of DNA methylation as a treatment in atherosclerosis is in its early infancy. The environmental factors which change the methylation pattern of DNA and also the molecular underlying mechanisms of atherogenesis need more investigations. Actually, there are many mechanisms which are implicated in DNA methylation. Further studies are required to illustrate the epigenetic complex network of atherosclerosis. Besides, the comprehensive knowledge about atherosclerotic underlying mechanisms, DNA methylation profile of genome may provide diagnostic biomarkers and ground-breaking therapies in the field of atherosclerosis.
Table 1. Atherosclerosis-related genes which are regulated through DNA methylation mechanism.
Antioxidants in atherosclerosis could have a therapeutic effect. As mentioned earlier, oxidative stress is linked with the development and formation of the atherosclerotic plaques. As a result, antioxidants could be a therapy for atherosclerosis patients. Generally, clinical trials on antioxidant therapy did not show any promises in case of atherosclerosis, though one of these antioxidants, probucol, inhibit ox-LDL, reduce the occurrence of vascular events and delay atherosclerosis progression. Vitamins, angiotensin-converting enzyme inhibitors (ACEI), calcium antagonists, angiotensin receptor antagonists, and statins can effectively suppress nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOXs) activity, decrease oxidative stress and eventually alleviate the atherosclerosis [Citation107].
Investigation on Decitabine (5-aza-dC) showed that this product could ameliorate atherosclerosis. 5-Aza-dC may apply its atheroprotective effects through antagonizing infiltration and activation of immune cells such as macrophage [Citation108]. This information could be used as a treatment approach for atherosclerosis.
In contrast to DNA mutations, epigenetic alterations such as DNA methylation are reversible and as such are targets for designing therapeutic approaches aimed at slowing the inevitable process of ageing.
Future direction of studies may involve potential targets of “epigenetic drugs”, such as DNMT inhibitors. Therefore, interventions linked to gene regulation through epigenetic may propose new possibilities to manage diseases in the near future.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Hansson GK, Robertson A-K, Söderberg-Nauclér C. Inflammation and atherosclerosis. Annu Rev Pathol. 2006;1:297–329.
- Singh M, Bedi US. Is atherosclerosis regression a realistic goal of statin therapy and what does that mean? Curr Atherosc Rep. 2013;15:294.
- Worthley SG, Osende JI, Helft G, et al. Coronary artery disease: pathogenesis and acute coronary syndromes. Mt Sinai J Med. 2001;68(3):167–181.
- Sipahi I, Tuzcu EM. Candidate mechanisms for regression of coronary atherosclerosis with high-dose statins. Am J Cardiovasc Drugs. 2008;8:365–371.
- Libby P, Ridker PM. Inflammation and atherothrombosis: from population biology and bench research to clinical practice. J Am Coll Cardiol. 2006;48:A33–A46.
- Študentová H, Indráková J, Petrová P, et al. Risk factors of atherosclerosis during systemic therapy targeting vascular endothelial growth factor. Oncol Lett. 2016;11:939–944.
- van Diepen JA, Berbée JF, Havekes LM, et al. Interactions between inflammation and lipid metabolism: relevance for efficacy of anti-inflammatory drugs in the treatment of atherosclerosis. Atherosclerosis. 2013;228:306–315.
- Bennett M, Wang J, Uryga AK, et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation. 2015;132:1909–1919.
- Yu X-H, Fu Y-C, Zhang D-W, et al. Foam cells in atherosclerosis. Clin Chim Acta. 2013;424:245–252.
- Sanjadi M, Rezvanie Sichanie Z, Totonchi H, et al. Atherosclerosis and autoimmunity: a growing relationship. Int J Rheum Dis. 2018;21:908–921.
- Zhang X, Fu R, Yu J, et al. DNA demethylation: where genetics meets epigenetics. Cpd. 2014;20:1625–1631.
- Fritz EL, Papavasiliou FN. Cytidine deaminases: AIDing DNA demethylation? Genes Dev. 2010;24:2107–2114.
- Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011;146:866–872.
- Needham BL, Smith JA, Zhao W, et al. Life course socioeconomic status and DNA methylation in genes related to stress reactivity and inflammation: the multi-ethnic study of atherosclerosis. Epigenetics. 2015;10:958–969.
- Aslani S, Mahmoudi M, Garshasbi M, et al. Evaluation of DNMT1 gene expression profile and methylation of its promoter region in patients with ankylosing spondylitis. Clin Rheumatol. 2016;35:2723–2731.
- Karami J, Mahmoudi M, Amirzargar A, et al. Promoter hypermethylation of BCL11B gene correlates with downregulation of gene transcription in ankylosing spondylitis patients. Genes Immun. 2017;18:170.
- Aslani S, Mahmoudi M, Karami J, et al. Epigenetic alterations underlying autoimmune diseases. Autoimmunity. 2016;49:69–83.
- Aslani S, Jafari N, Javan MR, et al. Epigenetic modifications and therapy in multiple sclerosis. Neuromol Med. 2017;19:11–23.
- Foma AM, Aslani S, Karami J, et al. Epigenetic involvement in etiopathogenesis and implications in treatment of systemic lupus erythematous. Inflamm Res. 2017;66:1057–1073.
- Zaina S, Lund G. Cardiovascular epigenome-wide association studies: is epigenetics falling short? Curr Opin Lipidol. 2014;25:474–475.
- Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204.
- Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315.
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282.
- Zhang Y, Zeng C. Role of DNA methylation in cardiovascular diseases. Clin Exp Hypertension. 2016;38:261–267.
- Bogdanović O, Veenstra G. DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma. 2009;118:549–565.
- Daniel FI, Cherubini K, Yurgel LS, et al. The role of epigenetic transcription repression and DNA methyltransferases in cancer. Cancer. 2011;117:677–687.
- Roman-Gomez J, Jimenez-Velasco A, Agirre X, et al. Promoter hypomethylation of the LINE-1 retrotransposable elements activates sense/antisense transcription and marks the progression of chronic myeloid leukemia. Oncogene. 2005;24:7213.
- Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36.
- Liu M, Timmons BW. The effect of acute exercise on neutrophil reactive oxygen species production and inflammatory markers in healthy prepubertal and adult males. Pediatr Exerc Sci. 2016;28:55–63.
- Kim GH, Ryan JJ, Marsboom G, et al. Epigenetic mechanisms of pulmonary hypertension. Pulm Circ. 2011;1:347–356.
- Kobayashi S, Inoue N, Azumi H, et al. Expressional changes of the vascular antioxidant system in atherosclerotic coronary arteries. Jat. 2002;9:184–190.
- Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313–20316.
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239.
- Nanduri J, Prabhakar NR. Epigenetic regulation of carotid body oxygen sensing: clinical implications. In: Peers C, Kumar P, Wyatt C, et al., editors. Arterial chemoreceptors in physiology and pathophysiology. Switzerland: Springer International Publishing; 2015. p. 1–8.
- Lim S-O, Gu J-M, Kim MS, et al. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: methylation of the E-cadherin promoter. Gastroenterology. 2008;135:2128–2140. e8.
- Niu Y, DesMarais TL, Tong Z, et al. Oxidative stress alters global histone modification and DNA methylation. Free Radic Biol Med. 2015;82:22–28.
- Afanas’ev I. Mechanisms of superoxide signaling in epigenetic processes: relation to aging and cancer. A&D. 2015;6:216.
- Weitzman SA, Turk PW, Milkowski DH, et al. Free radical adducts induce alterations in DNA cytosine methylation. Proc Natl Acad Sci. 1994;91:1261–1264.
- Turk PW, Laayoun A, Smith SS, et al. DNA adduct 8-hydroxyl-2'-deoxyguanosine (8-hydroxyguanine) affects function of human DNA methyltransferase. Carcinogenesis 1995;16:1253–1255.
- Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67:946–950.
- O'Hagan HM, Wang W, Sen S, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell. 2011;20:606–619.
- Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Curr Opin Pediatr. 2009;21:243.
- Valko M, Rhodes C, Moncol J, et al. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40.
- Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935.
- Shpyleva S, Ivanovsky S, de Conti A, et al. Cerebellar oxidative DNA damage and altered DNA methylation in the BTBR T + tf/J mouse model of autism and similarities with human post mortem cerebellum. PloS One. 2014;9:e113712.
- Wilson GA, Dhami P, Feber A, et al. Resources for methylome analysis suitable for gene knockout studies of potential epigenome modifiers. GigaScience 2012;1:3.
- Morgan GT, Jones P, Bellini M. Association of modified cytosines and the methylated DNA-binding protein MeCP2 with distinctive structural domains of lampbrush chromatin. Chromosome Res. 2012;20:925–942.
- Vilkaitis G, Merkiene E, Serva S, et al. The mechanism of DNA cytosine-5 methylation kinetic and mutational dissection of Hhai methyltransferase. J Biol Chem. 2001;276:20924–20934.
- Griffiths EA, Gore SD. MicroRNA: mIR-ly regulators of DNMT? Blood. 2009;113:6269–6270.
- Vanhoutte P, Shimokawa H, Feletou M, et al. Endothelial dysfunction and vascular disease – a 30th anniversary update. Acta Physiol (Oxf). 2017;219:22–96.
- Kuvikova I, Shevchuk S. Patients with have a level of endothelin-1 an antifosfolipid syndrome: connection is with motion of disease, disfunction of endothelia and atherosclerosis. Likars' ka Sprava. 2014;12:26–33.
- Buttery L, Springall D, Chester A, et al. Inducible nitric oxide synthase is present within human atherosclerotic lesions and promotes the formation and activity of peroxynitrite. Lab Invest. 1996;75:77–85.
- Sugiyama S, Okada Y, Sukhova GK, et al. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol. 2001;158:879–891.
- Ding Z, Liu S, Wang X, et al. Hemodynamic shear stress via ROS modulates PCSK9 expression in human vascular endothelial and smooth muscle cells and along the mouse aorta. Antioxid Redox Signal. 2015;22:760–771.
- Stenvinkel P, Karimi M, Johansson S, et al. Impact of inflammation on epigenetic DNA methylation–a novel risk factor for cardiovascular disease? J Intern Med. 2007;261:488–499.
- Shuto T, Furuta T, Oba M, et al. Promoter hypomethylation of Toll-like receptor-2 gene is associated with increased proinflammatory response toward bacterial peptidoglycan in cystic fibrosis bronchial epithelial cells. FASEB J. 2006;20:782–784.
- Karimi M, Johansson S, Stach D, et al. LUMA (LUminometric Methylation Assay)-a high throughput method to the analysis of genomic DNA methylation . Exp Cell Res. 2006;312:1989–1995.
- Ianni M, Porcellini E, Carbone I, et al. Genetic factors regulating inflammation and DNA methylation associated with prostate cancer. Prostate Cancer Prostatic Dis. 2013;16:56.
- Cipollone F, Fazia ML. COX-2 and atherosclerosis. J Cardiovasc Pharmacol. 2006;47:S26–S36.
- Denke MA, Grundy SM. Hypercholesterolemia in elderly persons: resolving the treatment dilemma. Ann Intern Med. 1990;112:780–792.
- Tanaka K, Masuda J, Imamura T, et al. A nation-wide study of atherosclerosis in infants, children and young adults in Japan. Atherosclerosis. 1988;72:143–156.
- Gallotta G, Iazzetta N, Milan G, et al. Prevalence of peripheral arterial disease in an elderly rural population of southern Italy. Gerontology. 1997;43:289–295.
- Holliday R. The inheritance of epigenetic defects. Science. 1987;238:163–170.
- Holliday R. The significance of DNA methylation in cellular aging. In: Woodhead AD, Blackett AD, Hollaender A, editors. Molecular biology of aging. New York, NY: Springer; 1985. p. 269–283.
- Wilson VL, Jones PA. DNA methylation decreases in aging but not in immortal cells. Science. 1983;220:1055–1057.
- Mays-Hoopes L, Chao W, et al. Decreased methylation of the major mouse long interspersed repeated DNA during aging and in myeloma cells. Dev Genet. 1986;7:65–73.
- Wilson VL, Smith R, Ma S, et al. Genomic 5-methyldeoxycytidine decreases with age. J Biol Chem. 1987;262:9948–9951.
- Drinkwater RD, Blake TJ, Morley AA, et al. Human lymphocytes aged in vivo have reduced levels of methylation in transcriptionally active and inactive DNA. Mutat Res DNAging. 1989;219:29–37.
- Issa J. CpG-island methylation in aging and cancer. Curr Top Microbiol Immunol. 2000;249:101–118.
- Post WS, Goldschmidt-Clermont PJ, Wilhide CC, et al. Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system. Cardiovasc Res. 1999;43:985–991.
- Ying AK, Hassanain HH, Roos CM, et al. Methylation of the estrogen receptor-α gene promoter is selectively increased in proliferating human aortic smooth muscle cells. Cardiovasc Res. 2000;46:172–179.
- Laukkanen MO, Mannermaa S, Hiltunen MO, et al. Local hypomethylation in atherosclerosis found in rabbit ec-sod gene. Arterioscler Thromb Vasc Biol. 1999;19:2171–2178.
- Steucke KE, Tracy PV, Hald ES, et al. Vascular smooth muscle cell functional contractility depends on extracellular mechanical properties. J Biomech. 2015;48:3044–3051.
- Krychtiuk KA, Kastl SP, Hofbauer SL, et al. Monocyte subset distribution in patients with stable atherosclerosis and elevated levels of lipoprotein (a). J Clin Lipidol. 2015;9:533–541.
- Berliner JA, Watson AD. A role for oxidized phospholipids in atherosclerosis. N Engl J Med. 2005;353:9–11.
- Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317.
- Sazonova OV, Isenberg BC, Herrmann J, et al. Extracellular matrix presentation modulates vascular smooth muscle cell mechanotransduction. Matrix Biol. 2015;41:36–43.
- Choi ET, Collins ET, Marine LA, et al. Matrix metalloproteinase-9 modulation by resident arterial cells is responsible for injury-induced accelerated atherosclerotic plaque development in apolipoprotein E–deficient mice. Atvb. 2005;25:1020–1025.
- Johnson JL, George SJ, Newby AC, et al. Divergent effects of matrix metalloproteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc Natl Acad Sci. 2005;102:15575–15580.
- Luttun A, Lutgens E, Manderveld A, et al. Loss of matrix metalloproteinase-9 or matrix metalloproteinase-12 protects apolipoprotein E-deficient mice against atherosclerotic media destruction but differentially affects plaque growth. Circulation. 2004;109:1408–1414.
- Jeong J-W, Kim JW, Ku SK, et al. Essential oils purified from Schisandrae semen inhibits tumor necrosis factor-α-induced matrix metalloproteinase-9 activation and migration of human aortic smooth muscle cells. BMC Complement Altern Med. 2015;15:7.
- Lacolley P, Regnault V, Nicoletti A, et al. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res. 2012;95:194–204.
- Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol. 2012;74:13–40.
- Spin JM, Maegdefessel L, Tsao PS. Vascular smooth muscle cell phenotypic plasticity: focus on chromatin remodelling. Cardiovasc Res. 2012;95:147–155.
- de Oca AM, Madueno JA, Martinez‐Moreno JM, et al. High‐phosphate‐induced calcification is related to SM22α promoter methylation in vascular smooth muscle cells. J Bone Miner Res. 2010;25:1996–2005.
- Hiltunen MO, Turunen MP, Häkkinen TP, et al. DNA hypomethylation and methyltransferase expression in atherosclerotic lesions. Vasc Med. 2002;7:5–11.
- Chen K-C, Wang Y-S, Hu C-Y, et al. OxLDL up-regulates microRNA-29b, leading to epigenetic modifications of MMP-2/MMP-9 genes: a novel mechanism for cardiovascular diseases. FASEB J. 2011;25:1718–1728.
- Little PJ, Rostam MA, Piva TJ, et al. Suramin inhibits PDGF‐stimulated receptor phosphorylation, proteoglycan synthesis and glycosaminoglycan hyperelongation in human vascular smooth muscle cells. J Pharm Pharmacol. 2013;65:1055–1063.
- Couffinhal T, Duplaa C, Moreau C, et al. Regulation of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 in human vascular smooth muscle cells. Circ Res. 1994;74:225–234.
- Azechi T, Sato F, Sudo R, et al. 5-Aza-2-deoxycytidine, a DNA methyltransferase inhibitor, facilitates the inorganic phosphorus-induced mineralization of vascular smooth muscle cells. J Atheroscler Thromb. 2014;21:463–476.
- Ning Y, Huang H, Dong Y, et al. 5-Aza-2'-deoxycytidine inhibited PDGF-induced rat airway smooth muscle cell phenotypic switching. Arch Toxicol. 2013;87:871–881.
- Perna AF, Ingrosso D. Atherosclerosis determinants in renal disease: how much is homocysteine involved? Nephrol Dial Transplant. 2016;31:860–863.
- Mandaviya PR, Stolk L, Heil SG. Homocysteine and DNA methylation: a review of animal and human literature. Mol Genet Metab. 2014;113:243–252.
- Ji C, Kaplowitz N. Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. Wjg. 2004;10:1699.
- Kaul S, Zadeh AA, Shah PK. Homocysteine hypothesis for atherothrombotic cardiovascular disease: not validated. J Am Coll Cardiol. 2006;48:914–923.
- Castro R, Rivera I, Struys EA, et al. Increased homocysteine and S-adenosylhomocysteine concentrations and DNA hypomethylation in vascular disease. Clin Chem. 2003;49:1292–1296.
- Yideng J, Jianzhong Z, Ying H, et al. Homocysteine-mediated expression of SAHH, DNMTs, MBD2, and DNA hypomethylation potential pathogenic mechanism in VSMCs. DNA Cell Biol. 2007;26:603–611.
- Jiang Y, Sun T, Xiong J, et al. Hyperhomocysteinemia‐mediated DNA hypomethylation and its potential epigenetic role in rats. Acta Biochim Biophys Sin. 2007;39:657–667.
- Han XB, Zhang HP, Cao CJ, et al. Aberrant DNA methylation of the PDGF gene in homocysteine-mediated VSMC proliferation and its underlying mechanism. Mol Med Rep. 2014;10:947–954.
- Huang Y-S, Zhi Y-F, Wang S-R. Hypermethylation of estrogen receptor-α gene in atheromatosis patients and its correlation with homocysteine. Pathophysiology. 2009;16:259–265.
- Leach NV, Dronca E, Vesa SC, et al. Serum homocysteine levels, oxidative stress and cardiovascular risk in non-alcoholic steatohepatitis. Eur J Int Med. 2014;25:762–767.
- Bao XmZheng H. Atorvastatin attenuates homocysteine‐induced migration of smooth muscle cells through mevalonate pathway involving reactive oxygen species and p38 MAPK. Clin Exp Pharmacol Physiol. 2015;42:865–873.
- Yideng J, Zhihong L, Jiantuan X, et al. Homocysteine-mediated PPARα, γ DNA methylation and its potential pathogenic mechanism in monocytes. DNA Cell Biol. 2008;27:143–150.
- Jamaluddin MS, Yang X, Wang H. Hyperhomocysteinemia, DNA methylation and vascular disease. Clinical Chemical Laboratory Medicine 2007;45:1660–1666.
- Yi-Deng J, Tao S, Hui-Ping Z, et al. Folate and ApoE DNA methylation induced by homocysteine in human monocytes. DNA Cell Biol. 2007;26:737–744.
- Wang J, Yu L, Zheng X. PTPα-mediated Src activation by EGF in human breast cancer cells. Acta Biochim Biophys Sin (Shanghai). 2013;45:320–329.
- Yang X, Li Y, Li Y, et al. Oxidative stress-mediated atherosclerosis: mechanisms and therapies. Front Physiol. 2017;8:600.
- Cao Q, Wang X, Jia L, et al. Inhibiting DNA methylation by 5-Aza-2'-deoxycytidine ameliorates atherosclerosis through suppressing macrophage inflammation. Endocrinology. 2014;155:4925–4938.
- Rasmussen MA, Holst B, Tümer Z, et al. Transient p53 suppression increases reprogramming of human fibroblasts without affecting apoptosis and DNA damage. Stem Cell Rep. 2014;3:404–413.
- Ma S, Zhang H, Sun W, et al. Hyperhomocysteinemia induces cardiac injury by up-regulation of p53-dependent Noxa and Bax expression through the p53 DNA methylation in ApoE−/− mice. Acta Biochim Biophys Sin. 2013;45:391–400.
- Hara S, Takano T, Fujikawa T, et al. Forced expression of DNA methyltransferases during oocyte growth accelerates the establishment of methylation imprints but not functional genomic imprinting. Human Mol Genet. 2014;23:3853–3864.
- Wang T, Chen M, Liu L, et al. Nicotine induced CpG methylation of Pax6 binding motif in StAR promoter reduces the gene expression and cortisol production. Toxicol Appl Pharmacol. 2011;257:328–337.
- Canani RB, Paparo L, Nocerino R, et al. Differences in DNA methylation profile of Th1 and Th2 cytokine genes are associated with tolerance acquisition in children with IgE-mediated cow’s milk allergy. Clin Epigenet. 2015;7:38.
- Joyce BT, Gao T, Liu L, et al. Longitudinal study of DNA methylation of inflammatory genes and cancer risk. Cancer Epidemiol Prevent Biomarkers 2015;24:1531–1538.
- Deaton AM, Cook PC, De Sousa D, et al. A unique DNA methylation signature defines a population of IFN‐γ/IL‐4 double‐positive T cells during helminth infection. Eur J Immunol. 2014;44:1835–1841.
- Wierda RJ, Kuipers HF, van Eggermond MC, et al. Epigenetic control of CCR5 transcript levels in immune cells and modulation by small molecules inhibitors. J Cell Mol Med. 2012;16:1866–1877.
- Kennedy A, Schmidt EM, Cribbs AP, et al. A novel upstream enhancer of FOXP3, sensitive to methylation‐induced silencing, exhibits dysregulated methylation in rheumatoid arthritis Treg cells. Eur J Immunol. 2014;44:2968–2978.
- Kim D, Kubzansky LD, Baccarelli A, et al. Psychological factors and DNA methylation of genes related to immune/inflammatory system markers: the VA Normative Aging Study. BMJ Open. 2016;6:e009790.
- Liu C, Xu D, Sjöberg J, et al. Transcriptional regulation of 15-lipoxygenase expression by promoter methylation. Exp Cell Res. 2004;297:61–67.
- Hastings NE, Simmers MB, McDonald OG, et al. Atherosclerosis-prone hemodynamics differentially regulates endothelial and smooth muscle cell phenotypes and promotes pro-inflammatory priming. Am J Physiol Cell Physiol. 2007;293:C1824–C33.
- Laukkanen MO, Mannermaa S, Hiltunen MO, et al. Local hypomethylation in atherosclerosis found in rabbit ec-sod gene. Arterioscler Thromb Vasc Biol. 1999;19:2171–2178.
- Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Curr Opin Pediatr. 2009;21:243.
- Carless MA, Kulkarni H, Kos MZ, et al. Genetic effects on DNA methylation and its potential relevance for obesity in Mexican Americans. PLoS One 2013;8:e73950.
- Chan Y, Fish JE, D'Abreo C, et al. The cell-specific expression of endothelial nitric oxide synthase: a role for DNA methylation. J Biol Chem. 2004;279:35087–35100.
- Ning Y, Huang H, Dong Y, et al. 5-Aza-2′-deoxycytidine inhibited PDGF-induced rat airway smooth muscle cell phenotypic switching. Arch Toxicol. 2013;87:871–881.
- Park J, Jang KL. Hepatitis C virus represses E-cadherin expression via DNA methylation to induce epithelial to mesenchymal transition in human hepatocytes. Biochem Biophys Res Commun. 2014;446:561–567.
- Liu W-b, Cui Z-h, Ao L, et al. Aberrant methylation accounts for cell adhesion-related gene silencing during 3-methylcholanthrene and diethylnitrosamine induced multistep rat lung carcinogenesis associated with overexpression of DNA methyltransferases 1 and 3a. Toxicol Appl Pharmacol. 2011;251:70–78.