
Abstract
Periodontitis refers to the inflammation of gums and the surrounding structures and caused by a bacterial infection. The infection occurs owing to poor oral hygiene and could destroy the bone and the gum over time if left untreated. The present study identified the involvement of a key long noncoding RNA (lncRNA), i.e. FGD5-AS1, in the pathogenesis of periodontitis by assessing its expression in the gingival tissues of patients diagnosed with chronic periodontitis (CP). Overexpression of FGD5-AS1 in primary human periodontal ligament cells (PDLCs) significantly reduced the lipopolysaccharide (LPS)-induced periodontitis, whereas its suppression aggravated this injury. Moreover, the miR-142-3p was markedly expressed in the gingival samples of patients diagnosed with CP and LPS-induced PDLCs. We found that the FGD5-AS1-mediated reduction in the inflammation was mediated through downside regulation of miR-142-3p, as evident from the upregulation of SOCS6, a target gene of miR-142-3p. Furthermore, the association between FGD5-AS1 and NF-κB pathway was detected. FGD5-AS1 was found to protect against LPS-stimulated PDLC injury through restraining the NF-κB signals. Based on these findings, we conclude that up-regulation of lncRNA FGD5-AS1 could protect against periodontitis via regulating the miR-142-3p/SOCS6/NF-κB signals. Therefore, the FGD5-AS1/miR-142-3p/SOCS6 axis may act as an important indicator in explaining the pathogenesis of periodontitis.
Introduction
Periodontitis is a common dental disease that is present worldwide. It is characterized by inflammatory responses to elevated levels of subgingival pathogens, causing damage to the soft tissues and alveolar bones supporting the teeth [Citation1,Citation2]. It is reported that mild to moderate periodontitis affects up to 50% of adults [Citation3]. However, severe periodontitis may raise the risks of systemic diseases, such as diabetes, rheumatoid arthritis, atherosclerosis and adverse pregnancy outcomes, thereby detrimentally affecting the systemic health of patients [Citation4–6]. Therefore, it is imperative to enhance the apprehensiveness of biological mechanisms underlying the pathogenesis of periodontitis to find better therapies.
Recently, endogenous long noncoding RNAs (lncRNAs) with more than 200 nucleotides have possessed extensive attention owing to their important roles in a large-scale biological processes [Citation7–9]. Moreover, an increasing number of studies have revealed several lncRNAs to be abnormally expressed in chronic periodontitis (CP), thus playing a crucial role in periodontitis pathogenesis [Citation10,Citation11]. However, the exact roles of differentially expressed key lncRNAs in periodontitis pathogenesis have not been completely understood. High-throughput RNA sequencing and microarray data on periodontitis have revealed the abnormal expression of lncRNA FGD5-AS1 to be associated with CP [Citation11]. However, these data require verification through physiological experiments including the role of lncRNA FGD5-AS1 and its impact of action.
In the current study, we determined the levels of FGD5-AS1 lncRNA in the gingival tissues of patients diagnosed with CP. Furthermore, to understand the possible mechanism of FGD5-AS1 in periodontitis, periodontal ligament cells (PDLCs) were isolated and treated with lipopolysaccharide (LPS) to induce periodontitis-caused inflammatory damage. The effects of abnormal expression of FGD5-AS1 on LPS-disposed PDLC injury were then evaluated by detecting cell viability, apoptosis, and production of inflammatory cytokines. Several studies have pointed out that lncRNAs serving as competitive endogenous RNAs (ceRNAs) and sponging miRNAs in various diseases [Citation12–14]. For example, FGD5-AS1 lncRNA, as a ceRNA, was proved to target miR-142-3p [Citation11]. Based on these data, we investigated whether FGD5-AS1 regulates LPS-induced PDLC injury via sponging miR-142-3p. Moreover, a regulatory pattern between miR-142-3p and suppressor of cytokine signalling 6 (SOCS6) was detected. Furthermore, NF-κB signals have been reported to be the central bridge involved in immune and inflammatory responses [Citation15,Citation16]. We, therefore, studied the association between FGD5-AS1 and NF-κB pathway. We believe that our discoveries would supply a theoretical basis for illuminating the pathogenesis of periodontitis.
Materials and methods
Gingival sample collection
Totally, 26 individuals suffered from CP and 20 healthy controls from April 2016 to December 2018 were chosen as subjects in this study. Gingival tissues from cases who were suffered from CP were collected surgically, followed by an original ineffectual nonsurgical scaling and root planning in accordance with the established professional and required oral hygiene of patients. Gingival tissues were also obtained while crown-lengthening routines from healthy individuals with following inclusion criteria: clinical attachment loss < 4 mm, probing depth (PD) < 4 mm, and no alveolar bone destruction at radiographic level. All collected gingival tissue samples were frozen in liquid nitrogen and then stored immediately at –80 °C for further experiments. Exclusion criteria included patients with smoking, rheumatoid arthritis, the appearance of any systemic disease, for instance, allergy type 1 and 2 diabetes, and uptake of drugs containing corticosteroids, and antibiotics within 1 month before the surgery. This study obtained the approval of the local ethics committee, and written consent was acquired from all enrolled individuals.
Cell culture and treatment
As previously described, we isolated PDLCs from the explanted healthy periodontal ligament in the middle third of the periodontal membrane root of impacted third molars [Citation17]. PDLCs were then cultured in DMEM (Dulbecco’s Modified Eagle Medium; Sangon Biotech; Shanghai, China) including 10% fetal bovine serum, 100,000 mg/mL streptomycin sulfate, 10,000 IU/mL penicillin G, 1% L-glutamine, and 25 mg/mL amphotericin B. Cells were cultivated at 37 °C with 5% CO2. After 80% confluency was achieved, PDLCs were treated with Porphyromonas gingivalis LPS (100 ng/mL; Sangon Biotech) for different time points.
Cell transfection
For cell transfection, PDLCs were further cultured into 6-well plates (2 × 105 cells/well), followed by transfection with pcDNA3.1, pc-FGD5-AS1, si-NC, si-FGD5-AS#1 or #2, miR-142-3p mimics, mimics NC, miR-142-3p inhibitor, inhibitor NC, pEX-SOCS6, pEX, sh-SOCS6 or sh-NC (GenePharma; Shanghai, China) using the Lipofectamine 2000 (Sangon Biotech) following the manufacturer’s instruction. After 48 h of cultivation, RT-qPCR was chosen for assessing the transfection efficiency.
Real-time qPCR
We used RNAiso Plus (Takara Bio; Tokyo, Japan) to extract the total RNA from both tissues and cells. The purified RNA was used for producing the complementary DNA (cDNA) by reverse-transcribed method using the PrimeScript RT Master Mix (for gene amplification, Perfect Real Time) or Mir-X miRNA First-Strand Synthesis Kit (for miRNA amplification, Takara). RT-qPCR assay was carried out by the SYBR® Premix Ex Taq™ II kit (Takara) for detecting the expression of target genes. The products were run on an ABI 7500 RT-PCR system (Bio Red). U6 and β-actin were used as references for miRNAs and RNAs, respectively. 2–ΔΔCt method was chosen for analyzing the relative expression standards of target genes.
MTT assay
We seeded the PDLCs into 96-well plates (1 × 104 cells/well). After various dispose, the obtained PDLCs in each well were mixed with 500 μg/mL of [3–(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide solution (MTT; Sigma-Aldrich) for further experiment. After 3 h of incubation, dimethyl sulfoxide solution (200 μL, Sigma-Aldrich) was mixed with cells in each well and mixed with the cells to melt the reaction product formazan. Finally, microplate reader (BioRad Model 550; Hercules, CA, USA) was chosen for detecting the absorbance of each well at 570 nm.
Apoptosis assay
PDLCs were harvested and washed in phosphate-buffered saline (PBS) for further use. The washed PDLCs were fixed in 70% ethanol. FITC Annexin V (50 μg/mL RNase A) (Sigma-Aldrich) was chosen for the subsequent staining. After 1 h incubation at room temperature darkly, FACScan system (BioRad) was used for assessment of the percentage of apoptotic cells. FlowJo software was used for analyzing the data.
Western blot
PDLCs were collected after different treatments and lysed in the cell lysis buffer (Sangon Biotech) to extract the total protein. The proteins in an equal amount (50 μg/lane) were resolved by 12% SDS-polyacrylamide gels, afterwards, the protein gel was transferred onto the prepared polyvinylidene fluoride (PVDF) membranes (Millipore; Billerica, MA, USA). Subsequently, 5% non-fat skim milk/TBST was chosen for blocking all PVDF membranes, then the target proteins were detected via incubating with primary antibodies at 4 °C overnight. The primary antibodies include Bax, Bcl-2, cleaved-caspase-3, pro-caspase-3, cleaved-caspase-9, pro-caspase-9, SOCS6, p65, IκBα, p-p65, p-IκBα, and β-actin (1:1,000 dilution; Abcam, Cambridge, UK). We choose β-actin for internal control. Finally, the membranes were incubated with appropriate secondary antibodies. The enhanced chemiluminescence reagent (Pierce; Rockford, IL, USA) was chosen for protein signals detection.
ELISA assay
Following the manufacturer’s protocol, the cytokine production in the supernatants of treated and untreated PDLCs was analyzed using the human IL-6, IL-8, IL-1β and TNF-α ELISA (Duoset kits; R&D Systems, USA). The optical density at 450 nm in each group was generated with 570 nm compensation.
Target prediction and luciferase reporter analysis
Based on the data obtained from TargetScan, SOCS6 was predicted and chosen as the latent target of miR-142-3p. To verify the prediction, we amplified the two kinds of types of (wild-type, WT; mutant, MUT) 3’-UTR regions in the SOCS6 sequences including the binding sequence of the putative miR-142-3p, and then inserted the amplified sequence into the pMIR GLO vector (Promega; Madison, WI, USA) at XhoI and XbaI restriction sites to produce the luciferase reporter plasmids (SOCS6-WT or SOCS6-MUT). PDLCs were then co-transfected with the following vectors, luciferase reporter plasmids (SOCS6-WT or SOCS6-MUT, 1 μg) and miR-142-3p vector (mimics or mimic NC, 50 pmoL) using the Lipofectamine 2000 transfection reagent. At 48 h of post-transfection, dual-luciferase activity assay system (Sangon Biotech) was chosen for the assessment of the luciferase activity of each luciferase reporter plasmid in each group. The Renilla luciferase activity was chosen as the internal control.
Statistical analysis
Three independent experiments were conducted for each assay. All values are calculated and expressed as mean values ± standard deviations (SDs). Significance was analyzed using one-way analysis of variance (ANOVA) or a two-tailed Student’s t-test. p < .05 was chosen as of significance.
Results
FGD5-AS1 is poorly expressed in gingival samples of patients diagnosed with CP
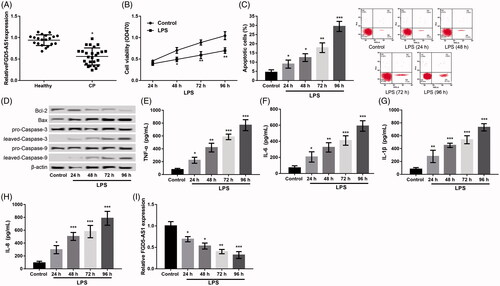
Our results indicated that the expression of FGD5-AS1 was dramatically low in the gingival samples of patients diagnosed with CP in comparison with that in the gingival samples obtained from healthy controls (p < .05, ). These findings indicated that FGD5-AS1 participates in the development of CP.
Figure 1. FGD5-AS1 was poorly expressed in gingival samples of patients with CP (CP) and lipopolysaccharide (LPS) induced injury in PDLCs; and FGD5-AS1 was poorly expressed in LPS-stimulated PDLCs. (A) FGD5-AS1 was poorly expressed in gingival samples of patients with CP (CP). *p < .05 compared with healthy controls. (B) MTT assay showing the cell viability at different time points after LPS treatment. (C) Flow cytometry showing the cell apoptosis at different time points after LPS treatment. (D) Western blot showing the expression of apoptosis-related proteins at different time points after LPS treatment. (E–H) ELISA showing the production of inflammatory cytokines including TNF-α, IL-6, IL-1β and IL-8 at different time points after LPS treatment. (I) qPCR showing the expression of FGD5-AS1 at different time points after LPS treatment. The experiments were performed three times and data are expressed in terms of mean ± SD. *p < .05, **p < .01, and ***p < .001 compared with control.
FGD5-AS1 level is decreased in LPS-stimulated PDLCs
As mentioned in the methods section, PDLCs were treated with LPS for different time points to generate the periodontitis cell model. Relative to the control, LPS dispose significantly reduced the cell viability (); induced cell apoptosis (); increased the levels of apoptotic proteins including Bax/Bcl-2, cleaved/pro-caspase-3 and cleaved/pro-caspase-9; but accelerated the levels of inflammatory cytokines of TNF-α, IL-6, IL-1β and IL-8 () in a time-dependent way (p < .05). These results implied that LPS induced inflammatory damage in PDLCs. Furthermore, the expression of FGD5-AS1 was also found to decrease significantly in PDLCs following LPS dispose in a time-dependent way (p < 0.05, ).
Up-regulation of FGD5-AS1 alleviates LPS-stimulated PDLC injury, whereas suppression of FGD5-AS1 aggravates this injury
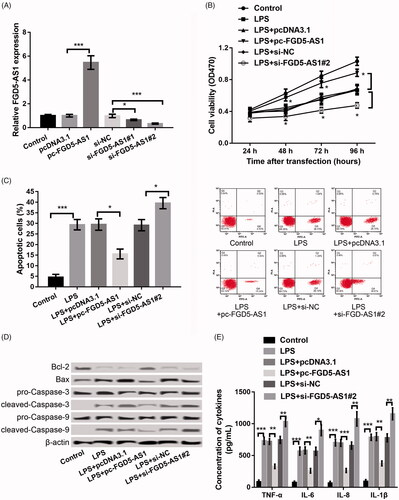
To explore the role of FGD5-AS1 in CP, FGD5-AS1 was both overexpressed and suppressed in PDLCs to detect the effect of its abnormal expression on cell physiology including viability, apoptosis, and cytokine secretion in LPS-disposed PDLCs. As revealed in , FGD5-AS1 expression was dramatically increased in the pc-FGD5-AS1 transfection group. However, it was remarkably decreased in si-FGD5-AS1#1 and #2 transfection groups (p < 0.05), indicating high transfection efficiency. The si-FGD5-AS1#2 was used in the following experiments owing to the stronger inhibitory efficiency than that of si-FGD5-AS1#1 (). Further experiments showed that overexpression of FGD5-AS1 in LPS-stimulated PDLCs significantly enhanced the cell viability (); inhibited apoptosis (); decreased the levels of apoptotic proteins including Bax/Bcl-2, cleaved/pro-caspase-3 and cleaved/pro-caspase-9 (); and cut down the secretion of inflammatory cytokines of TNF-α, IL-6, IL-1β and IL-8 (), suggesting that overexpression of FGD5-AS1 alleviated the LPS-induced PDLC injury. As expected, its suppression led to reverse effects (p < .05).
Figure 2. Overexpression of FGD5-AS1 alleviated LPS-induced PDLC injury, whereas suppression of FGD5-AS1 aggravated this injury. (A) qPCR showing the expression of FGD5-AS1 after transfection with pc-FGD5-AS1, si-FGD5-AS1#1 or #2 and the corresponding controls, respectively. (B) MTT assay showing the cell viability after different treatments. (C) Flow cytometry showing cell apoptosis after different treatments. (D) Western blot showing the expression of apoptosis-related proteins after different treatments. (E) ELISA showing the production of inflammatory cytokines including TNF-α, IL-6, IL-1β, and IL-8 after different treatments. The experiments were performed three times and data are expressed in terms of mean ± SD. *p < .05, **p < .01, and ***p < .001 compared with the corresponding control.
FGD5-AS1 reversed miR-142-3p and suppression of FGD5-AS1 aggravates LPS-induced cell injury by adding miR-142-3p
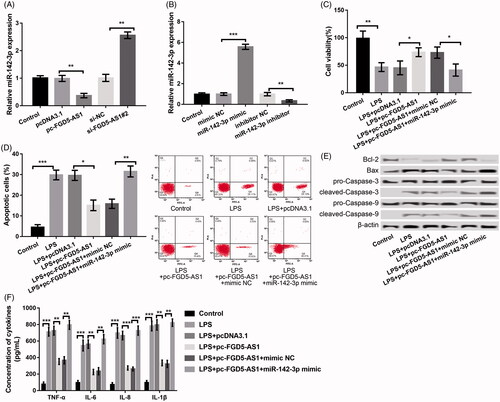
We found that miR-142-3p was decreased in the pc-FGD5-AS1 group relative to its expression in the pcDNA3.1 control group (p < .01). It was also markedly increased in the si-FGD5-AS1#2 group as compared to that in the sh-NC group (p < .01) (), deducting the presence of a negative regulation between FGD5-AS1 and miR-142-3p. miR-142-3p was successfully overexpressed/inhibited in PDLCs by transfecting with miR-142-3p mimic/inhibitor, respectively (p < .01, ). Afterwards, the jointed impacts of overexpression of FGD5-AS1 and miR-142-3p on LPS-induced PDLC injury were investigated. In relation to LPS + pc-FGD5-AS1 + mimic NC group, co-transfection of pc-FGD5-AS1 and miR-142-3p markedly changeover the influences of pc-FGD5-AS1 alone on LPS-induced cell injury in the LPS-disposed PDLCs. These impacts were mediated through decreasing cell viability (); promoting cell apoptosis (); accelerating the expression levels of Bax/Bcl-2, cleaved/pro-caspase-3 and cleaved/pro-caspase-9 (); and inducing the secretion of inflammatory factors of TNF-α, IL-6, IL-1β and IL-8 () (p < .05).
Figure 3. FGD5-AS1 negatively regulated the expression of miR-142-3p and suppression of FGD5-AS1 alleviated LPS-induced cell injury by negative regulation of miR-142-3p. (A) qPCR showing the expression of miR-142-3p after transfection with pc-FGD5-AS1, si-FGD5-AS1#2 and corresponding controls, respectively. (B) qPCR showing the expression of miR-142-3p after transfection with miR-142-3p mimic, miR-142-3p inhibitor and corresponding controls, respectively. (C) MTT assay showing cell viability after different treatments. (D) Flow cytometry showing cell apoptosis after different treatments. (E) Western blot showing the expression of apoptosis-related proteins after different treatments. (F) ELISA showing the production of inflammatory cytokines including TNF-α, IL-6, IL-1β and IL-8 after different treatments. The experiments were performed three times and data are expressed in terms of mean ± SD. *p < .05, **p < .01, and ***p < .001 compared with corresponding control.
SOCS6 is targeted by miR-142-3p
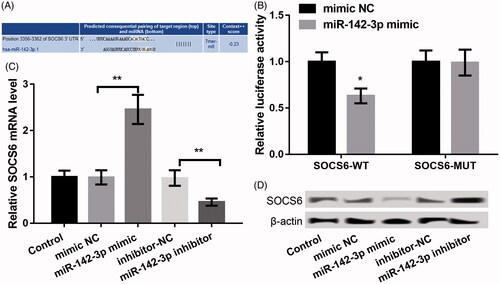
A downside expression of SOCS6 was discovered in periodontal disease [Citation18]. Also, the targeted evidence about miR-142-3p vs. SOCS6 was reported in CD4+ T cells of multiple sclerosis [Citation19] and in nasopharyngeal carcinoma [Citation20]. We discovered that SOCS6 was a possible target of miR-142-3p (http://www.targetscan.org/cgi-bin/targetscan/vert_71/) using TargetScan (version 7.1). Their complementary pairing sequences are shown in . Moreover, only the luciferase activity of SOCS6-WT was markedly decreased after co-transfection of miR-142-3p mimic relative to mimic NC (p < .05, ), suggesting SOCS6 is targeted by miR-142-3p. In addition, the data revealed that SOCS6 levels in the miR-142-3p mimic group were markedly reduced relative to that in the mimic NC group, whereas its expression in the miR-142-3p inhibitor group was markedly higher compared with the inhibitor NC group (p < .01, ), confirming SOCS6 is targeted by miR-142-3p.
Figure 4. SOCS6 was identified as a target gene of miR-142-3p. (A) The complementary pairing sequence between miR-142-3p and SOCS6. (B) Luciferase reporter assay displaying the luciferase activity of SOCS6-WT and SOCS6-MUT after cotransfection with miR-142-3p mimic or mimic NC. (C–D) qPCR and western blot showing the mRNA and protein expression of SOCS6 after transfection with miR-142-3p mimic, miR-142-3p inhibitor and the corresponding controls, respectively. The experiments were performed three times and data are expressed in terms of mean ± SD. *p < .05, and **p < .01 compared with the corresponding controls.
Influences of miR-142-3p on LPS-induced PDLC injury are through targeting of SOCS6
We next analyzed the influences of miR-142-3p and SOCS6 in CP. miR-142-3p was remarkably up-regulated in gingival samples of patients diagnosed with CP (p < .01, ) and LPS-induced PDLCs in a time-dependent way (p < .05, ). On the contrary and for obvious reasons, the expression of SOCS6 was down-regulated (p < 0.05, ). Transfection with pEX-SOCS6 and sh-SOCS6 led to successful expression and knock-down of SOCS6 in PDLCs, respectively (p < .01, ). Furthermore, experiments demonstrated that inhibition of miR-142-3p significantly accelerated the LPS-disposed cell injury in PDLCs through enhancing the cell viability (); inhibiting apoptosis (); decreasing the levels of Bax/Bcl-2, cleaved/pro-caspase-3 and cleaved/pro-caspase-9 (); and suppressing the secretion of inflammatory factors, including TNF-α, IL-6, IL-1β and IL-8 () (p < .05). However, these effects were remarkably changeover after transfection of miR-142-3p inhibitor and si-SOCS6 synchronously in LPS-stimulated PDLCs. These data indicated that miR-142-3p could aggravate LPS-resulted injury in PDLCs by targeting SOCS6.
Figure 5. Effects of miR-142-3p on LPS-induced PDLC injury demonstrated through targeting SOCS6. (A,B) qPCR showing the expression of miR-142-3p in the gingival samples of patients with CP and LPS-induced PDLCs. (C,D) qPCR and western blot showing the expression of SOCS6 in gingival samples of patients with CP and LPS-induced PDLCs. (E) qPCR and western blot showing the expression of SOCS6 after transfection with pEX-SOCS6, sh-SOCS6 and the corresponding controls, respectively. (F) MTT assay showing the cell viability after different treatments. (G) Flow cytometry showing cell apoptosis after different treatments. (H) Western blot showing the expression of apoptosis-related proteins after different treatments. (I) ELISA showing the production of inflammatory cytokines including TNF-α, IL-6, IL-1β and IL-8 after different treatments. (J) Western blot showing the protein expression levels of p/t-p65 and p/t-IκBα expression in FHCs that were treated with PDLCs and co-transfected with pc-FGD5-AS1, miR-142-3p mimic, pEX-SOCS6 and/or their respective controls. (K) The regulatory mechanism chart of FGD5-AS1 in regulating LPS-induced PDLC injury. The experiments were performed three times and data are expressed in terms of mean values ± SD. *p < .05 and **p < .01, compared with the respective controls.
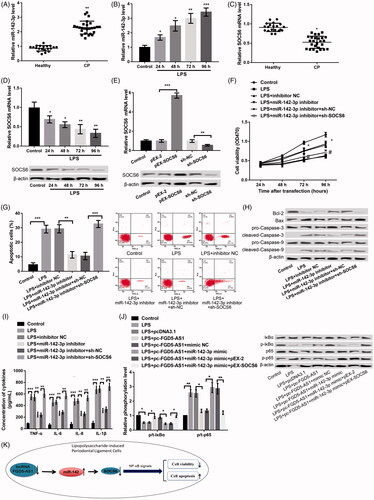
FGD5-AS1 protects against LPS-induces PDLCs injury through inhibiting NF-κB signals
We further explored the regulatory pattern between FGD5-AS1 and NF-κB signals in LPS-stimulated PDLCs (). Our results revealed that LPS treatment markedly augmented the level of p/t-p65 but cut down the level of p/t-IκBα, indicating the LPS-mediated activation of the NF-κB signals. Overexpression of FGD5-AS1 significantly decreased p/t-p65 and increased p/t-IκBα expression in LPS-stimulated PDLCs, indicating that overexpression of FGD5-AS1 could reverse the LPS-induced activation of NF-κB signal. Moreover, simultaneous overexpression of FGD5-AS1 and miR-142-3p could reverse the influences of overexpression of FGD5-AS1 alone on activating the NF-κB signals; these effects were further changeover after co-transfection of pc-SOCS6 ().
Discussion
Numerous studies have highlighted the regulatory effects of lncRNAs in several human diseases [Citation21–23]. However, crucial lncRNAs that are known to participate in the development of diseases comprise only the tip of the iceberg. In this current research, our team explored the expression and effects of FGD5-AS1 in the pathogenesis of periodontitis. Our research revealed that FGD5-AS1 was down-regulated in patients with CP and LPS-disposed PDLCs. Moreover, overexpression of FGD5-AS1 alleviated the LPS-disposed PDLC injury, whereas its suppression aggravated this injury. Furthermore, FGD5-AS1 negatively regulated miR-142-3p, whereas si-FGD5-AS1 alleviated the LPS-induced cell injury via negatively regulating miR-142-3p. In addition, SOCS6 was targeted directly by miR-142-3p, and it was revealed that miR-142-3p alleviated the LPS-induced injury in PDLCs by targeting SOCS6. Another significant discovered result of the present study was that FGD5-AS1 protected against LPS-disposed PDLCs injury through depressing the NF-κB signals. Nevertheless, these data merit further discussion.
There are growing studies that suggest lncRNAs to play a pivotal role in disease pathogenesis by acting as ceRNAs to modulate the gene expression through sponging miRNAs [Citation24,Citation25]. Previous evidence pointed out that miR-142-3p is targeted by FGD5-AS1 as a part of the ceRNA network [Citation11]. Moreover, miR-142-3p is considered as a mediator of inflammation and functions in regulating proinflammatory signalling pathways [Citation26,Citation27]. Previous evidence has confirmed that miR-142-3p gets dysregulated in several inflammatory diseases, such as keratitis [Citation28], psoriasis [Citation29], ulcerative colitis [Citation30], osteoarthritis [Citation31] and dermatitis [Citation32]. Notably, Perri et al. demonstrated that miR-142-3p was markedly added in periodontitis [Citation33], suggesting its potential role in periodontitis development. In the current study, we found that FGD5-AS1 negatively regulated miR-142-3p and its suppression enhanced LPS-disposed cell injury via up-regulation of miR-142-3p. Given the key role of miR-142-3p in inflammation, we believe that FGD5-AS1 could hold back the development of periodontitis via negatively regulating the miR-142-3p.
In addition, SOCS6 has been discovered as targeted by miR-142-3p. SOCS6 is a member of SOCS family proteins that are known as regulators of inflammatory responses induced by bacterial infections [Citation34]. Also, SOCS6 is identified as a cytokine-inducible negative regulator of cytokine signalling [Citation35] and is involved in negative regulation of receptor signalling and inducing apoptosis by targeting mitochondrial proteins [Citation36]. Evidence also points out that SOCS6 is implicated in diverse human cancers [Citation37,Citation38]. In the present research, SOCS6 level was discovered as very low in gingival samples of patients with CP and LPS-disposed PDLCs. Moreover, the influences of inhibition of miR-142-3p on LPS-resulted cell injury in LPS-resulted PDLCs were remarkably reversed after simultaneous inhibition of miR-142-3p and knocking down of SOCS6. Although the role of SOCS6 in periodontitis has not been deciphered, based on the association between SOCS6 and cytokine signalling, we speculate that the si-SOCS6 could accelerate the development of periodontitis and miR-142-3p may prevent periodontitis by targeting SOCS6.
Furthermore, the NF-κB signal has been proved to be the central mediator that regulates inflammation, periodontitis, and atherogenesis [Citation39]. Nakajima et al. demonstrated that anandamide could block LPS-triggered activation of NF-κB signalling and consequently regulated hyperinflammatory reactions in periodontitis [Citation40]. Similarly, Dong et al. revealed that osteopontin promoted bone destruction in periapical periodontitis via activating the NF-κB signal [Citation40]. In the present study, overexpression of FGD5-AS1 could changeover the LPS-resulted activation of NF-κB signal. Moreover, simultaneous overexpression of FGD5-AS1 and miR-142-3p could reverse the influences of overexpression of FGD5-AS1 alone on activating NF-κB signal that was further changeover after co-transfection with pc-SOCS6. We thus hypothesize that FGD5-AS1 could prevent the development of periodontitis through regulating the miR-142-3p/SOCS6 axis so as to realize the suppressed effects on activating NF-κB signals.
In conclusion, the results of the current study reported a down-regulation of FGD5-AS1 in patients with CP. Up-regulation of RNA FGD5-AS1 could protect against periodontitis via regulating the miR-142-3p/SOCS6/NF-κB pathway. Thus, the FGD5-AS1/miR-142-3p/SOCS6 axis may provide a promising tactics for the treatment of periodontitis.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Hajishengallis G. Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends Immunol. 2014;35:3–11.
- Ratheesh V, Subramanian S, Prakash PSG, et al. Evaluation of association of vitamin D receptor genetic polymorphism with severe CP in an ethnic Tamilian population. Genet Test Mol Biomarkers. 2018;22:615–621.
- Eberhard J, Jepsen S, Jervøe‐Storm PM, et al. Full‐mouth treatment modalities (within 24 hours) for CP in adults. Cochrane Database Syst Rev. 2015;4:CD004622.
- Genco RJ, Van Dyke TE. Prevention: reducing the risk of CVD in patients with periodontitis. Nat Rev Cardiol. 2010;7:479–480.
- Lalla E, Papapanou PN. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol. 2011;7:738–748.
- Madianos PN, Bobetsis YA, Offenbacher S. Adverse pregnancy outcomes (APOs) and periodontal disease: pathogenic mechanisms. J Clin Periodontol. 2013;40:S170–S180.
- Kanduri C. Long noncoding RNAs: Lessons from genomic imprinting. Biochim Biophys Acta. 2016;1859:102–111.
- Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–1307.
- Iyer MK, Niknafs YS, Malik R, et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015;47:199.
- Zou Y, Li C, Shu F, et al. lncRNA expression signatures in periodontitis revealed by microarray: the potential role of lncRNAs in periodontitis pathogenesis. J Cell Biochem. 2015;116:640–647.
- Li S, Liu X, Li H, et al. Integrated analysis of long noncoding RNA-associated competing endogenous RNA network in periodontitis. J Periodont Res. 2018;53:495–505.
- Salmena L, Poliseno L, Tay Y, et al. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358.
- Qi X, Zhang D, Wu N, et al. ceRNA in cancer: possible functions and clinical implications. J Med Genet. 2015;52:710–718.
- Du H, Chen Y. Competing endogenous RNA networks in cervical cancer: function, mechanism, and perspective. J Drug Target. 2018;1–47.
- Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733.
- Zhang H, Sun SC. NF-κB in inflammation and renal diseases. Cell Biosci. 2015;5:63
- Li C, Yue J, Huang X, et al. miR-21 and miR-101 regulate PLAP-1 expression in periodontal ligament cells. Mol Med Rep. 2012;5:1340.
- Moffatt CE, Lamont RJ. Porphyromonas gingivalis induction of microRNA-203 expression controls suppressor of cytokine signaling 3 in gingival epithelial cells. Infect Immun. 2011;79:2632–2637.
- Sanders KA, Benton MC, Lea RA, et al. Next-generation sequencing reveals broad down-regulation of microRNAs in secondary progressive multiple sclerosis CD4+ T cells. Clin Epigenet. 2016;8:87–96.
- Qi X, Li J, Zhou C, et al. MiR-142-3p suppresses SOCS6 expression and promotes cell proliferation in nasopharyngeal carcinoma. Cell Physiol Biochem. 2015;36:1743–1752.
- Bhan A, Soleimani M, Mandal SS. Long noncoding RNA and cancer: a new paradigm. Cancer Res. 2017;77:3965–3981.
- Delás MJ, Hannon GJ. lncRNAs in development and disease: from functions to mechanisms. Open Biol. 2017;7:170121.
- Simion V, Haemmig S, Feinberg MW. LncRNAs in vascular biology and disease. Vascul Pharmacol. 2019;114:145–156.
- Xu J, Xu Y. The lncRNA MEG3 downregulation leads to osteoarthritis progression via miR-16/SMAD7 axis. Cell Biosci. 2017;7:69.
- Dai L, Zhang G, Cheng Z, et al. Knockdown of LncRNA MALAT1 contributes to the suppression of inflammatory responses by up-regulating miR-146a in LPS-induced acute lung injury. Connect Tissue Res. 2018;59:581–592.
- Yuan Z, Luo G, Li X, et al. PPARγ inhibits HMGB1 expression through upregulation of miR-142-3p in vitro and in vivo. Cell Signal. 2016;28:158–164.
- Xu G, Zhang Z, Wei J, et al. microR-142-3p down-regulates IRAK-1 in response to Mycobacterium bovis BCG infection in macrophages. Tuberculosis. 2013;93:606–611.
- Boomiraj H, Mohankumar V, Lalitha P, et al. Human corneal MicroRNA expression profile in fungal keratitis. Invest Ophthalmol Vis Sci. 2015;56:7939.
- Pivarcsi A, Meisgen F, Xu N, et al. Changes in the level of serum microRNAs in patients with psoriasis after antitumour necrosis factor-α therapy. Br J Dermatol. 2013;169:563–570.
- Schaefer JS, Attumi T, Opekun AR, et al. MicroRNA signatures differentiate Crohn’s disease from ulcerative colitis. BMC Immunol. 2015;16:5.
- Wang X, Guo Y, Wang C, et al. MicroRNA-142-3p inhibits chondrocyte apoptosis and inflammation in osteoarthritis by targeting HMGB1. Inflammation. 2016;39:1718–1728.
- Ulrik R, Lise L, Thomas L, et al. MicroRNA expression in early mycosis fungoides is distinctly different from atopic dermatitis and advanced cutaneous T-cell lymphoma. Anticancer Res. 2014;34:7207–7217.
- Perri R, Nares S, Zhang S, et al. MicroRNA modulation in obesity and periodontitis. J Dent Res. 2012;91:33–38.
- Duncan SA, Baganizi DR, Sahu R, et al. SOCS proteins as regulators of inflammatory responses induced by bacterial infections: a review. Front Microbiol. 2017;8:2431.
- Liu Z, Xie D, Zhang H. Long noncoding RNA neuroblastoma-associated transcript 1 gene inhibits malignant cellular phenotypes of bladder cancer through miR-21/SOCS6 axis. Cell Death Dis. 2018;9:1042.
- Kabir NN, Sun J, Rönnstrand L, et al. SOCS6 is a selective suppressor of receptor tyrosine kinase signaling. Tumor Biol. 2014;35:10581–10589.
- Yuan D, Wang W, Su J, et al. SOCS6 functions as a tumor suppressor by inducing apoptosis and inhibiting angiogenesis in human prostate cancer. Curr Cancer Drug Targets. 2018;18:894–904.
- Cheng L, Kong B, Zhao Y, et al. miR-494 inhibits cervical cancer cell proliferation through upregulation of SOCS6 expression. Oncol Lett. 2018;15:3075–3080.
- Nichols TC, Fischer TH, Deliargyris EN, et al. Role of nuclear factor‐kappa B (NF‐κB) in inflammation, periodontitis, and atherogenesis. Ann Periodontol. 2001;6:20–29.
- Nakajima Y, Furuichi Y, Biswas KK, et al. Endocannabinoid, anandamide in gingival tissue regulates the periodontal inflammation through NF-kappaB pathway inhibition. FEBS Lett. 2006;580:613–619.