
Abstract
The morphological feature of apoptosis is induced by oxygen and glucose deprivation (OGD) in cardiomyocytes H9c2 cells. Salvianolic acid B (Sal-B) has been studied in several pathological progresses, whereas it is still unclear whether maternally expressed gene 3 (MEG3) is an intermediate regulator during this progress. After pre-incubation with Sal-B and stimulation with OGD, viability and apoptosis of were examined in MEG3-overexpressed H9c2 cells. Cyclin D1, apoptosis-correlated proteins and regulators of signalling pathways were quantified with Western blot assay. MEG3 was detected by quantitative reverse transcription PCR (qRT-PCR). Sal-B was implicated in the enhancement of cell viability and suppression of apoptosis in OGD-treated H9c2 cells by repressing MEG3. In addition, MEG3 overexpression exerted an inhibitory effect on murine double minute 2 (MDM2) expression while aggrandized p53 expression in OGD-treated H9c2 cells which were pre-incubated with Sal-B. Furthermore, MEG3 overexpression abolished the up-regulative effect of Sal-B on phosphorylation of adenosine monophosphate-activated protein kinase (AMPK) in OGD-treated H9c2 cells. These results indicated that cardio-protective function of Sal-B might be ascribed to its down-regulatory property on MEG3 expression which hence blocks p53 and triggers AMPK activation in OGD-treated cells.
Keywords:
Introduction
Myocardial ischemia is a serious stress scenario which results in the morphological characteristics of apoptosis. Glucose deprivation causes oxidative stress evidenced by induction of reactive oxygen species and decrease of glutathione level in cultured cardiac myocytes [Citation1]. An essential mechanism has been elaborated that endoplasmic reticulum generates and propagates apoptotic signals, such as preceding the activation of caspase [Citation2]. Furthermore, caspase-3 and caspase-9 were processed into the active forms accompanied by the release of cytochrome c, suggesting that ischemia activates the mitochondrial apoptosis pathway in cardiac myocytes [Citation3]. In the study of apoptotic mechanism, oxygen and glucose deprivation (OGD) are normally applied to mimic the pathological characteristics of ischemia.
A growing number of bioactive compounds, such as tanshinone, ginsenoside Rb and salidroside, have been discovered to exert cytoprotective capability against OGD-caused apoptosis by regulating apoptosis-associated signalling pathways [Citation4–6]. Analogously, salvianolic acid B (Sal-B; its chemical structure as shown in ), as the main functional phenolic component obtained from the root of Salvia miltiorrhiza Bunge [Citation7], has been reported to possess a cardio-protective effect against multifarious stress-induced damage. The possible mechanism, that Sal-B mediates the activation or blockage of cellular signalling pathways, might be responsible for its anti-apoptotic and pro-proliferative function [Citation8–10].
Figure 1. Chemical structure of salvianolic acid B. Molecular formula: C36H30O16; Average mass: 718.61.
Human homologue maternally expressed gene 3 (MEG3), as a conserved long non-coding RNA, was identified as an imprinting gene and mapped on human chromosome 14q [Citation11]. It is defined as a crucial mediator of cellular functions. Additionally, it is implicated in the development of cardiac fibrosis and diastolic dysfunction after cardiac stress [Citation12]. A recent study found that the methylation of MEG3 promoter region is altered by curcumin and then its expression is enhanced in hepatocellular cancer [Citation13]. Accordingly, we considered that a MEG3-associated mechanism, through which Sal-B improved the tolerance of myocardial cells against OGD, might be subsistent.
Furthermore, it has been demonstrated that the MEG3 mediates ischemic neuronal death by interacting with p53 in stroke [Citation14]. MEG3 induces p53 accumulation and accordingly causes the down-regulation of murine double minute 2 (MDM2) at least in part [Citation15]. In addition, adenosine monophosphate-activated protein kinase (AMPK) plays a critical effect on the transcriptional induction and phosphorylation of p53 during the induction of apoptosis under carbon source deprivation [Citation16]. These regulators are involved in the apoptosis-related signalling pathways. Since the active component has been found to regulate MEG3 in pathological process [Citation13], we were thereafter motivated to explore whether Sal-B is able to modulate these signalling pathways through regulating MEG3.
In order to warrant these speculations, we established an in vitro H9c2 cells model based on their vulnerability to OGD. Subsequently, we pre-incubated H9c2 cells with Sal-B before OGD stimulation. Thereafter, MEG3-transfected H9c2 cells were applied to prove that Sal-B exhibited a cardio-protective activity by attenuating MEG3 expression and secondly mediated MDM2/p53 and AMPK signalling pathways.
Materials and methods
Cell culture, OGD stimulation and Sal-B pre-incubation
H9c2 cells were purchased from American Type Culture Collection (ATCC; Rockville, MD, USA). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma, St. Louis, MO, USA) containing 10% fetal bovine serum (v/v) (FBS; Gibco, Gaithersburg, MD, USA), 100 U/mL penicillin (Sigma) and 100 μg/mL streptomycin (Sigma) at 37 °C in an atmosphere of 95% air and 5% CO2. For OGD stimulation, H9c2 cells were cultured in a glucose-free DMEM in an anaerobic and temperature controlled (37 ± 0.5 °C) chamber which were flushed with 5% CO2 and 95% N2 for various time intervals. The glucose-free medium was replaced with normal medium after OGD for 2 h and H9c2 cells were continually cultured for 24 h. H9c2 cells maintained in the normal medium served as control. Sal-B was purchased from Paipai (Guangzhou, China). H9c2 cells were pre-incubated with Sal-B at different concentrations (1, 5, and 10 μM) for 24 h. Sal-B was prepared with phosphate buffer solution (PBS; Invitrogen, Carlsbad, CA, USA).
Cell viability assay
After pre-treated with or without Sal-B and stimulated with OGD, the viability of H9c2 cells was investigated with a cell counting kit-8 (CCK-8; Dojindo Molecular Technologies, Gaithersburg, MD). In brief, CCK-8 solution was added into the culture medium after stimulation and the cultures were incubated for 1 h at 37 °C in humidified 95% air and 5% CO2. The absorbance was measured at 450 nm using a Microplate Reader (Bio-Rad, Hercules, CA).
Apoptosis examination
Apoptotic cells were observed with a flow cytometry after staining with Annexin V-fluorescein isothiocyanate/propidium iodide (FITC/PI) apoptosis detection kit (Beijing Biosea Biotechnology, Beijing, China). Shortly, H9c2 cells were seeded in 6-well plates at a density of 100,000 cells per well. Successively, apoptotic cells and necrotic cells were stained according to the manufacturer’s instruction and then observed with a flow cytometer (Beckman Coulter, USA).
MEG3 overexpression and quantification
The full-sequence of MEG3 was constructed into pEX-2 plasmid (GenePharma; Shanghai, China) named as pMEG3. Lipofectamine 3000 reagent (Life Technologies Corporation) was applied to transfect pMEG3 into H9c2 cells for up-regulating MEG3 with reference to the manufacturer’s instruction. For verifying the transfection efficiency, quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analysis was performed to quantify the expression of MEG3. Primarily, total RNA was extracted from MEG3-transfected H9c2 cells using TRIzol reagent (Invitrogen) and DNase I (Promega, Madison, WI, USA). Reverse transcription was performed with the Mutiscribe RT and RNase Inhibitor Mix (Applied Biosystems, CA, USA). MEG3 was quantified using PCR with specific primers as described previously [Citation17]. Quantitative PCR data were normalized with GAPDH expression.
Western blot assay
RIPA Lysis buffer (Beyotime Biotechnology, Shanghai, China) combined with protease inhibitors (Roche, Guangzhou, China) was applied to extract the total proteins for Western blot assay. The concentration of total proteins was quantified with BCA™ protein assay kit (Pierce, Appleton, WI, USA). After separation with sodium dodecylsulphate-polyacrylamide gel electrophoresis (SDS-PAGE), proteins were transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Temecula, CA, USA). Consecutively, membranes were incubated with primary antibodies against cyclin D1 (ab16663; 1:100), Bax (ab32503; 1:1000) (all purchased from Abcam, Cambridge, MA, USA), caspase-3 (14220; 1:1000), caspase-9 (9508; 1:1000), MDM2 (3521; 1:1000), p53 (32532; 1:1000), total (t)-AMPK (5832; 1:1000), phosphor (p)-AMPK (50081; 1:1000) and β-actin (4967; 1:1000) (all obtained from Cell Signaling Technology, Danvers, MA, USA) overnight at 4 °C. Subsequently, the secondary antibodies, including rabbit anti-mouse (58802) and goat anti-rabbit (7074) (all obtained from Cell Signaling Technology) linked to horseradish peroxidase (HRP), specifically bound with the primary antibodies. Then, membranes carrying blots and antibodies were transferred onto the Bio-Rad ChemiDoc™ XRS system. The intensity of protein signals was quantified with Image Lab™ software (Bio-Rad, Shanghai, China).
Statistical analysis
The data from at least three individual experiments were presented as mean ± standard deviations (SD). p Values were calculated with Student’s t-test or one-way analysis of variance (ANOVA) followed by Dunnett test for comparing the difference. GraphPad Prism 6.0 was employed to perform statistical analysis. The significance was considered when p < .05.
Results
OGD-induced H9c2 damage
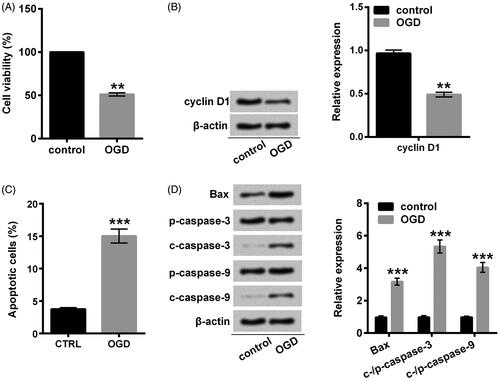
For investigating whether Sal-B protects H9c2 cells against OGD-induced injury, we established the OGD model with H9c2 cells by using the glucose deprivation and hypoxia conditions. Impairment of cell viability and advancement of apoptosis implied that we efficaciously constructed the OGD-induced injury models (p < .01 or p < .001; ). The obviously down-regulated cyclin D1 suggested that OGD retarded the progression of proliferation into S-phase since it has been demonstrated that cyclin D1, as a critical target of proliferative signal in G1 is required at S-phase in the cell cycle [Citation18]. Our results showed that cyclin D1 was down-regulated by OGD at protein level (p < .01; ), indicating that OGD impeded cell proliferation cycle. An antecedent study has revealed that ectopic expression of Bax induces apoptosis preceding caspase activation [Citation19]. Under OGD conditions, Bax overexpression, as well as cleavage of caspase-3 and caspase-9, were detected in our study (p < .001; ), suggesting that OGD committed H9c2 cells to apoptosis.
Figure 2. OGD induced H9c2 cells damage. (A) Cell viability was assessed with CCK-8. (B) A Western blot of cyclin D1 was represented with normalization to β-actin. (C) Apoptotic cells were observed with a flow cytometry after stain with Annexin V-FITC/PI. (D) Western blots of apoptosis-related proteins were suggested with normalization to β-actin. H9c2 cells were stimulated with OGD for 2 h. **p < .01 or ***p < .001 compared with the normoxia control. OGD: oxygen and glucose deprivation; CCK-8: cell counting kit-8; FITC/PI: fluorescein isothiocyanate/propidium iodide.
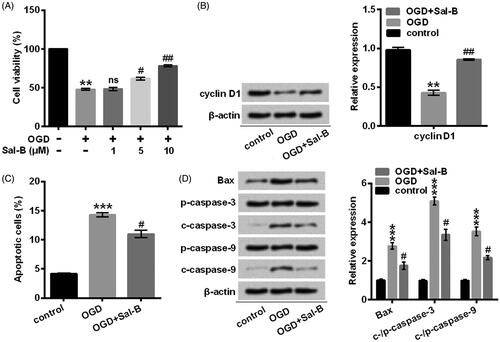
Sal-B administration moderated OGD-induced H9c2 damage
Afterwards, we pre-incubated H9c2 cells with Sal-B at the indicated concentrations (0, 1, 5, and 10 μM) before OGD stimulation. Our results showed that the Sal-B caused an improvement of cell viability in H9c2 cells subjected to OGD in a dose-dependent manner (p < .05 or p < .01; ). Nevertheless, Sal-B did not affect cell viability at a low concentration (p > .05; ). As a consequence, we pre-treated H9c2 cells with Sal-B at a concentration of 10 μM in the following experiments. In vitro studies of the advancement of cell viability and restraint of apoptosis provided unambiguous evidence that Sal-B displayed a protective function on the activities of H9c2 cells against OGD-induced damage (p < .05 or p < .01; ). The precise mechanism responsible for its protective effects appeared to be associated with the up-regulated expression of cyclin D1 (p < .01) (), down-regulated expression of Bax (p < .05) as well as the cleaved production of caspase-3 and caspase-9 (p < .05) (). Our abovementioned results revealed that Sal-B was implicated in the promotion of cell viability and inhibition of apoptosis through mediating factors of cell cycle and apoptosis.
Figure 3. Sal-B protected H9c2 cells against OGD-induced damage. (A) Cell viability was assessed with CCK-8. H9c2 cells were stimulated with OGD for 2 h before pre-incubation with Sal-B at indicated concentrations. (B) A Western blot of cyclin D1 was represented with normalization to β-actin. (C) Apoptotic cells were observed with a flow cytometry after stain with Annexin V-FITC/PI. (D) Western blots of apoptosis-related proteins were suggested with normalization to β-actin. H9c2 cells were stimulated with OGD for 2 h before pre-incubation with Sal-B (10 μM) for 24 h. ns: p > .05, **p < 0.01 or ***p < .001 compared with the normoxia control. #p < .05 or ##p < .01 compared with the OGD-treated group. Sal-B: salvianolic acid B; OGD: oxygen and glucose deprivation; CCK-8: cell counting kit-8; FITC/PI: fluorescein isothiocyanate/propidium iodide; ns: not significant; c: cleaved; p: pro.
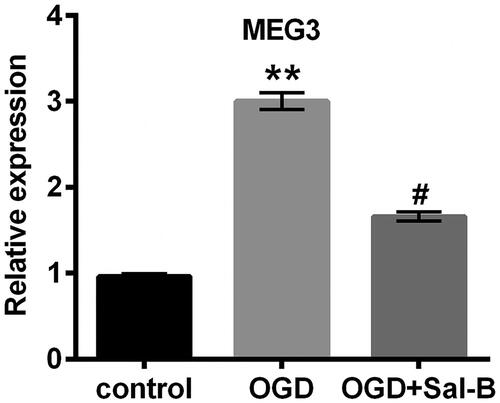
OGD-induced MEG3 overexpression was suppressed by Sal-B
The decreased generation of MEG3 has been examined in non-small cell lung cancer tumour tissues and its overexpression induced apoptosis and impeded cell viability [Citation17]. Furthermore, the increase in MEG3 expression was induced by hypoxia in H9c2 cells and the knockdown of MEG3 relieved hypoxia-induced injury [Citation20]. Analogously, the overexpression of MEG3 distinctly exhibited in OGD-treated H9c2 cells (p < .01; ). Interestingly, we found that Sal-B markedly abated the overexpression of MEG3 triggered by OGD (p < .05; ).
Figure 4. OGD-induced MEG3 overexpression was repressed by Sal-B. Relative expression of MEG3 was detected with qRT-PCR method in H9c2 cells stimulated with or without OGD for 2 h and/or pre-incubated with Sal-B (10 μM) for 2 h. **p < .01 compared with the normoxia control. #p < .05 compared with the OGD-treated group. MEG3: maternally expressed gene 3; OGD: oxygen and glucose deprivation; Sal-B: salvianolic acid B; qRT-PCR: quantitative reverse transcription PCR.
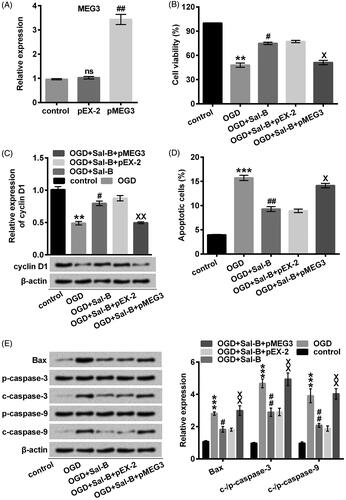
Down-regulation of MEG3 accounted for the pro-proliferative and anti-apoptotic functions of Sal-B
Our observation, that the obvious reduction of MEG3 by Sal-B would be a consequence of cytoprotection, stimulated us to fully elucidate whether MEG3 overexpression abrogated the protective functions of Sal-B against OGD-induced damage. Accordingly, we effectually elevated MEG3 by transfection (p < .01; ). Moreover, the cell viability was signally eliminated in MEG3-transfected H9c2 cells after pre-incubation with Sal-B and stimulation with OGD compared with the un-transfected cells (p < .05; ). The memorable decrease of cyclin D1 in MEG3-transfected H9c2 cells (p < .01; ) strongly suggested that Sal-B facilitated proliferation via inhibiting MEG3 expression induced by OGD. Besides, MEG3 overexpression noticeably abolished the anti-apoptotic effects of Sal-B in OGD-treated H9c2 cells (p < .05; ), in good agreement with the overexpression of Bax as well as the biosynthesis of cleaved caspase-3 and caspase-9 (all p < .01; ). These findings supported our notion that the effect of Sal-B would be a result of its negative modulation on MEG3 expression.
Figure 5. MEG3 overexpression abolished the protective effects of Sal-B against OGD-induced H9c2 cells damage. (A) MEG3 was determined with qRT-PCR after transfection with pMEG3 or not. ns: p > .05 compared with the normoxia control. ##p < .01 compared with the OGD-treated group. (B) Cell viability was assessed with CCK-8. H9c2 cells were stimulated with OGD for 2 h before pre-incubation with Sal-B at indicated concentrations. (C) A Western blot of cyclin D1 was represented with normalization to β-actin. (D) Apoptotic cells were observed with a flow cytometry after stain with Annexin V-FITC/PI. (E) Western blots of apoptosis-related proteins were suggested with normalization to β-actin. H9c2 cells were stimulated with OGD for 2 h before pre-incubation with Sal-B (10 μM) for 24 h. ns: p > .05, **p < .01 or ***p < .001 compared with the normoxia control. #p < .05 or ##p < .01 compared with the OGD-treated group. Xp < .05 or XXp < .01 compared with the OGD + Sal-B + pEX-2 group. CCK-8: cell counting kit-8; FITC/PI: fluorescein isothiocyanate/propidium iodide; MEG3: maternally expressed gene 3; OGD: oxygen and glucose deprivation; Sal-B: salvianolic acid B; MEG3: maternally expressed gene 3; ns: not significant; OGD: oxygen and glucose deprivation; Sal-B: salvianolic acid B; c: cleaved; p: pro; qRT-PCR: quantitative reverse transcription PCR.
Sal-B regulated MDM2/p53 and AMPK signalling pathways via MEG3
MEG3 transcription is required for p53 which in turn modulates proliferation and apoptosis [Citation17]. Additionally, the rapid degradation of p53 coincides with maximal MDM2 induction [Citation21]. Actually, MDM2 loss and p53 generation caused by OGD (p < .01 or p < .001; ) were thought to be necessary for the suppression of cell viability, disarrangement of proliferation and aggravation of apoptosis. It should be mentioned that Sal-B pre-incubation prevented the OGD-mediated MDM2 depletion and p53 generation (p < .01 or p < .001; ). Overexpression of MEG3 appeared to exert a conspicuously contrary influence on the expression of MDM2 and p53 under OGD and Sal-B situations compared with the un-transfected group (p < .01 or p < .001; ).
Figure 6. MEG3 overexpression reversed the modulatory effects of Sal-B on MDM2/p53 and AMPK signaling pathways in OGD-treated H9c2 cells. (A) MDM2, p53, (B) t-AMPK and p-AMPK were quantified with Western blot assay. H9c2 cells were stimulated with OGD or not for 2 h before pre-incubation with or without Sal-B (10 μM) for 24 h. *p < .05, **p < .01 or ***p < .001 compared with the normoxia control. ##p < .01 or ###p < .001 compared with the OGD-treated group. XXp < .01 or XXXp < .001 compared with the OGD + Sal-B + pEX-2 group. MEG3: maternally expressed gene 3; MDM2: murine double minute 2; AMPK: adenosine monophosphate activated protein kinase; OGD: oxygen and glucose deprivation; Sal-B: salvianolic acid B; p: phosphor; t: total.
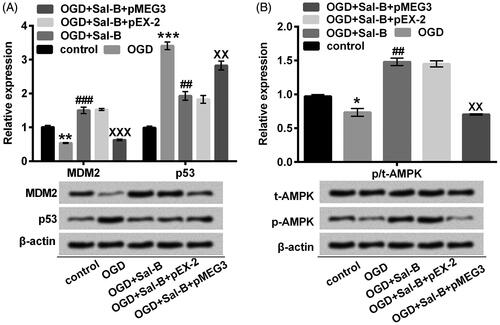
Through inducing the endogenous raptor phosphorylation, AMPK activation dictates a metabolic checkpoint controlling growth arrest and apoptosis in response to energy stress [Citation22]. As expected, the decreased phosphorylation of AMPK dramatically contributed the susceptibility of H9c2 cells to OGD-induced injury (p < .05; ). However, the activation of AMPK by Sal-B (p < .01; ) would deploy a potentially protective mechanism which may be associated with MEG3. This view was supported by our observation that Sal-B synergetic with transfected MEG3 did not overtly aggrandize the phosphorylated expression of AMPK (p < .01; ).
Discussion
A recent study indicated that Sal-B improved the structure and function of left ventricle by maintaining the integrity of mitochondria and nucleus and then repressing apoptosis process [Citation10]. Furthermore, the mechanistic investigation has been performed to address its cardio-protective function [Citation9,Citation23,Citation24]. Unfortunately, the MEG3-relative mechanism responsible for OGD-induced damage is still unclear. Considering that MEG3 was up-regulated in response to environmental stress [Citation20,Citation25], we established an in vitro OGD model to mimic the in vivo myocardial ischemia model for investigating the cyto-protective function of Sal-B and then anatomized the underlying mechanism.
Substantially, we established the in vitro OGD model to mimic the metabolic characteristic of ischemia by oxygen plus glucose deprivation. Our results showed that short-period OGD resulted in an increase of apoptosis and decrease of cell viability whereas Sal-B enabled H9c2 cells to tolerate OGD with the enhanced cell viability and impaired apoptosis. Above results demonstrated the cardio-protective function of Sal-B in accordance with previous in vitro and in vivo studies [Citation23,Citation26,Citation27]. It should be emphasized that the blockade of cyclin D1 synthesis was abolished by Sal-B in OGD-treated H9c2 cells, revealing that Sal-B possessed a pro-proliferative property. Moreover, our discovery that the expression of Bax and cleavage of caspase-3 and caspase-9 further illustrated the anti-apoptotic effects of Sal-B. A growing number of interconnecting pharmacological mechanisms might be involved, for instance, free-radical scavenging activity, modulated phosphorylation of signalling regulators, and transcriptional regulative function as mentioned by a reported review [Citation28]. Nonetheless, it was still unexplored whether MEG3 might be responsible for the cardio-protective property of Sal-B. We, therefore, studied its possibly molecular mechanisms.
Because MEG3 has been considered as a cell death promoter in the mediation of ischemic insults [Citation14], up-regulated MEG3 might exert an essential role in OGD-induced damage. As confirmed in a previous study, the aberrantly elevated expression of MEG3 has been observed in hypoxia-treated H9c2 cells and its knockdown alleviates hypoxia-induced injury [Citation20]. Similar results have been manifested in cerebral ischemic infarct of mice and hypoxia-treated neurons [Citation29]. Consistently, our study showed that ODG significantly elevated the expression of MEG3 in H9c2 cells. Furthermore, a down-regulated expression of MEG3 was observed in H9c2 cells which were pretreated with Sal-B and stimulated with ODG. Accordingly, we were stimulated to speculate that Sal-B played a positive effect on cardio-protection via suppressing MEG3. Based on the demonstrations, our assumption was valid. Although the potentially biochemical mechanisms are still indistinct, the correlatively biological processes seem to participate, such as Ca2+ influx [Citation30], RNA-DNA triplex structures formation [Citation31] and protease activity [Citation32].
Indeed, MEG3 has been demonstrated to correlate with cell proliferation in multiple cancers [Citation33,Citation34]. The accurate mechanistic study has been continuously performed. As a competing endogenous RNA for microRNA-181b, MEG3 knockdown contributes to an attenuation of apoptosis induced by hypoxia [Citation29]. The activation or inactivation of several signalling pathways is accompanied by the MEG3 overexpression or knockdown [Citation35,Citation36]. Under certain pathological conditions, MEG3 interacts with p53 protein for activating p53-mediated transcriptional activity and therewith mediates the expression of partial p53 target genes [Citation37]. In response to nutrient deprivation, MDM2 binds ribosomal proteins and then induces p53-mediated energy metabolism [Citation38]. Actually, hypoxia promotes p53 accumulation through repressing MDM2 expression [Citation39]. As a consequence, we concluded that down-regulated MEG3 served as an intermediary factor in the pro-proliferative and anti-apoptotic functions of Sal-B by mediating MDM/p53 signalling pathway. AMPK is another essential signalling regulator which is normally activated for alleviating apoptosis [Citation40,Citation41]. Our study implied that Sal-B coordinated OGD with AMPK phosphorylation by down-regulating MEG3.
In summary, we proved that H9c2 cells, pre-incubated with Sal-B, were more tolerant of OGD episode than un-incubated cells. In addition, we addressed that the cardio-protective function of Sal-B was ascribed to its down-regulatory property on MEG3 expression and hence to block p53 activation and trigger AMPK.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Marambio P, Toro B, Sanhueza C, et al. Glucose deprivation causes oxidative stress and stimulates aggresome formation and autophagy in cultured cardiac myocytes. Biochim Biophys Acta. 2010;1802:509–518.
- Szegezdi E, Duffy A, O’Mahoney ME, et al. ER stress contributes to ischemia-induced cardiomyocyte apoptosis. Biochem Biophys Res Commun. 2006;349:1406–1411.
- Bialik S, Cryns VL, Drincic A, et al. The mitochondrial apoptotic pathway is activated by serum and glucose deprivation in cardiac myocytes. Circ Res. 1999;85:403–414.
- Zheng K, Sheng Z, Li Y, et al. Salidroside inhibits oxygen glucose deprivation (OGD)/re-oxygenation-induced H9c2 cell necrosis through activating of Akt-Nrf2 signaling. Biochem Biophys Res Commun. 2014;451:79–85.
- Zhu JR, Tao YF, Lou S, et al. Protective effects of ginsenoside Rb(3) on oxygen and glucose deprivation-induced ischemic injury in PC12 cells. Acta Pharmacol Sin. 2010;31:273–280.
- Wu WY, Wang WY, Ma YL, et al. Sodium tanshinone IIA silate inhibits oxygen-glucose deprivation/recovery-induced cardiomyocyte apoptosis via suppression of the NF-kappaB/TNF-alpha pathway. Br J Pharmacol. 2013;169:1058–1071.
- Dong J, Liu Y, Liang Z, et al. Investigation on ultrasound-assisted extraction of salvianolic acid B from Salvia miltiorrhiza root. Ultrason Sonochem. 2010;17:61–65.
- Wang J, Zhang Y, Guo LL, et al. Salvianolic acid B inhibits the TLR4-NFkappaB-TNFalpha pathway and attenuates neonatal rat cardiomyocyte injury induced by lipopolysaccharide. Chin J Integr Med. 2011;17:775–779.
- Wang M, Sun GB, Sun X, et al. Cardioprotective effect of salvianolic acid B against arsenic trioxide-induced injury in cardiac H9c2 cells via the PI3K/Akt signal pathway. Toxicol Lett. 2013;216:100–107.
- Xu L, Deng Y, Feng L, et al. Cardio-protection of salvianolic acid B through inhibition of apoptosis network. PloS One. 2011;6:e24036.
- Miyoshi N, Wagatsuma H, Wakana S, et al. Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells. 2000;5:211–220.
- Piccoli MT, Gupta SK, Viereck J, et al. Inhibition of the cardiac fibroblast-enriched lncRNA Meg3 prevents cardiac fibrosis and diastolic dysfunction. Circ Res. 2017;121:575–583.
- Zamani M, Sadeghizadeh M, Behmanesh M, et al. Dendrosomal curcumin increases expression of the long non-coding RNA gene MEG3 via up-regulation of epi-miRs in hepatocellular cancer. Phytomedicine. 2015;22:961–967.
- Yan H, Yuan J, Gao L, et al. Long noncoding RNA MEG3 activation of p53 mediates ischemic neuronal death in stroke. Neuroscience. 2016;337:191–199.
- Zhou Y, Zhong Y, Wang Y, et al. Activation of p53 by MEG3 non-coding RNA. J Biol Chem. 2007;282:24731–24742.
- Okoshi R, Ozaki T, Yamamoto H, et al. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J Biol Chem. 2008;283:3979–3987.
- Lu KH, Li W, Liu XH, et al. Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer. 2013;13:461.
- Baldin V, Lukas J, Marcote MJ, et al. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7:812–821.
- Finucane DM, Bossy-Wetzel E, Waterhouse NJ, et al. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J Biol Chem. 1999;274:2225–2233.
- Gong L, Xu H, Chang H, et al. Knockdown of long non-coding RNA MEG3 protects H9c2 cells from hypoxia-induced injury by targeting microRNA-183. J Cell Biochem. 2018;119:1429–1440. Feb
- Haupt Y, Maya R, Kazaz A, et al. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299.
- Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226.
- Pan C, Lou L, Huo Y, et al. Salvianolic acid B and tanshinone IIA attenuate myocardial ischemia injury in mice by NO production through multiple pathways. Ther Adv Cardiovasc Dis. 2011;5:99–111.
- Xue L, Wu Z, Ji XP, et al. Effect and mechanism of salvianolic acid B on the myocardial ischemia-reperfusion injury in rats. Asian Pacific J Trop Med. 2014;7:280–284.
- Li X, Jun X, Biao X. GW29-e1836 Long non-coding RNA MEG3 knockdown attenuates endoplasmic reticulum stress-mediated apoptosis by targeting p53 after myocardial infarction. J Am Coll Cardiol. 2018;72:C63.
- Deng Y, Yang M, Xu F, et al. Combined salvianolic acid B and ginsenoside Rg1 exerts cardioprotection against ischemia/reperfusion injury in rats. PloS One. 2015;10:e0135435.
- Xu T, Wu X, Chen Q, et al. The anti-apoptotic and cardioprotective effects of salvianolic acid a on rat cardiomyocytes following ischemia/reperfusion by DUSP-mediated regulation of the ERK1/2/JNK pathway. PloS One. 2014;9:e102292.
- Wang J, Xiong X, Feng B. Cardiovascular effects of salvianolic Acid B. Evid Based Complement Alternat Med. 2013;2013:247948.
- Liu X, Hou L, Huang W, et al. The mechanism of long non-coding RNA MEG3 for neurons apoptosis caused by hypoxia: mediated by miR-181b-12/15-LOX signaling pathway. Front Cell Neurosci. 2016;10:201.
- Lam FF, Yeung JH, Kwan YW, et al. Salvianolic acid B, an aqueous component of Danshen (Salvia miltiorrhiza), relaxes rat coronary artery by inhibition of calcium channels. Eur J Pharmacol. 2006;553:240–245.
- Mondal T, Subhash S, Vaid R, et al. MEG3 long noncoding RNA regulates the TGF-beta pathway genes through formation of RNA-DNA triplex structures. Nat Commun. 2015;6:7743.
- Joe Y, Zheng M, Kim HJ, et al. Salvianolic acid B exerts vasoprotective effects through the modulation of heme oxygenase-1 and arginase activities. J Pharm Exp Ther. 2012;341:850–858.
- Kruer TL, Dougherty SM, Reynolds L, et al. Expression of the lncRNA maternally expressed gene 3 (MEG3) contributes to the control of lung cancer cell proliferation by the Rb pathway. PloS One. 2016;11:e0166363.
- Iyer S, Modali SD, Agarwal SK. Long noncoding RNA MEG3 is an epigenetic determinant of oncogenic signaling in functional pancreatic neuroendocrine tumor cells. Mol Cell Biol. 2017;37:e00278-17.
- Xia Y, He Z, Liu B, et al. Downregulation of Meg3 enhances cisplatin resistance of lung cancer cells through activation of the WNT/beta-catenin signaling pathway. Mol Med Rep. 2015;12:4530–4537.
- Yang NQ, Luo XJ, Zhang J, et al. Crosstalk between Meg3 and miR-1297 regulates growth of testicular germ cell tumor through PTEN/PI3K/AKT pathway. Am J Transl Res. 2016;8:1091–1099.
- Zhu J, Liu S, Ye F, et al. Long noncoding RNA MEG3 interacts with p53 protein and regulates partial p53 target Genes in hepatoma cells. PloS One. 2015;10:e0139790.
- Liu Y, He Y, Jin A, et al. Ribosomal protein-Mdm2-p53 pathway coordinates nutrient stress with lipid metabolism by regulating MCD and promoting fatty acid oxidation. Proc Natl Acad Sci USA. 2014;111:E2414–22.
- Alarcon R, Koumenis C, Geyer RK, et al. Hypoxia induces p53 accumulation through MDM2 down-regulation and inhibition of E6-mediated degradation. Cancer Res. 1999;59:6046–6051.
- Chen K, Li G, Geng F, et al. Berberine reduces ischemia/reperfusion-induced myocardial apoptosis via activating AMPK and PI3K-Akt signaling in diabetic rats. Apoptosis. 2014;19:946–957.
- Kambara T, Ohashi K, Shibata R, et al. CTRP9 protein protects against myocardial injury following ischemia-reperfusion through AMP-activated protein kinase (AMPK)-dependent mechanism. J Biol Chem. 2012;287:18965–18973.