
Abstract
Double-strand break (DSB) repair foci are important therapeutic targets. Here we describe platforms for delivery of macromolecules, nanomaterials and nanomedicines to repair foci. The strategy is based on the high affinity of the human 53BP1 protein for modified chromatin present at sites of DNA damage. As proof of concept, we created, expressed, and purified an engineered fragment of 53BP1 and coupled it to fluorescent streptavidin, a model cargo with no intrinsic affinity for repair foci. This binary complex was in turn coupled to the iron carrier protein, transferrin, which engages a high-affinity cell surface receptor. In a different version of the complex, transferrin was omitted and a protein transduction domain was incorporated directly into the primary structure of the 53BP1. These complexes were efficiently taken up into human osteosarcoma cells and synchronously released from endocytic vesicles by brief exposure to far-red light in the presence of the photosensitizer, disulfonated aluminum phthalocyanine. Upon release, the streptavidin cargo entered the nucleus and was recruited to repair foci. 53BP1-based platforms provide a method for targeted, temporally controlled delivery of macromolecular agents to sites of double-strand break repair. With the delivery platforms, we are capable to visualize, modify and redirect DSB repair pathways by coupling various nanomaterials to study machinery or manipulate for therapy purpose in the future.
Introduction
A long-term goal of the nanomedicine field is to understand cellular nanomachines and redirect their activity for therapeutic purposes. DNA double-strand break (DSB) repair machines are a prime example. DSB repair is one of the very few natural processes that permit targeted modification of chromosomal DNA sequences. The ability to visualize, modify and redirect these repair pathways would be useful in a variety of settings. These include the delivery of bright stable fluorophores to help visualize the repair process, the delivery of peptides or other molecules to inhibit repair in the context of radiation therapy, and the modification of pathway choice to favor homologous recombination (HR) in the context of gene correction therapy. Several macromolecular agents that inhibit DSB repair complex assembly or function have already been described, including aptamers, peptides and an antibody fragment ([Citation1–3], reviewed in [Citation4]). DSB repair is closely related with the sensitivity of radiation therapy and chemotherapy for cancer cells, DSB repair machinery could also be manipulated as a promising therapeutic target for cancer, like the poly ADP-ribose polymerase (PARP) inhibitor for ovarian cancer [Citation5] and breast cancer [Citation6]. Therefore, it is a valuable tool to establish an efficient platform for introducing macromolecular agent or therapeutic drugs to the DSB foci to study composition and function of these foci, as well as target the DNA breaks with various modifying substances for the treatment of cancer.
Attempts to target repair foci with macromolecular agents suffer from a common limitation, which that foci are relatively inaccessible. To reach them, a macromolecular agent must traverse the plasma membrane, evade cytoplasmic trapping, and enter the cell nucleus. In the present study, we explore a combination of components and technologies to systematically surmount these barriers and achieve delivery of a protein-based nano complex to repair foci. Studies here focus on the development of platforms for direct delivery of proteins and other materials, in contrast to the alternative approach of delivering a cDNA expression construct encoding the molecule of interest. Direct delivery has distinct advantages, including spatiotemporal control and the ability to deliver both natural substances such as proteins and synthetic material.
In the work described here, traversal of the plasma membrane was achieved by harnessing cellular endocytic pathways. Prior work has shown that conjugation to transferrin and folate can be used to promote endocytic uptake of proteins and other materials in a variety of cell types [Citation7,Citation8]. These ligands engage high-affinity surface receptors, which are internalized to the endosomal compartment, and the reducing environment in endosomes facilitate cleavage of a scissile (“self-immolating”) disulfide linker allows separation of ligand-receptor complex, which is recycled to the cell surface, and macromolecular cargo, which is retained within the cell [Citation9]. Receptor-mediated delivery is attractive because of its demonstrated potential for clinical translation [Citation8]. A related approach is based on protein transduction domains (PTD), which promote endocytic uptake by a non-receptor dependent pathway. PTDs have also been used to promote uptake in a variety of cell types (reviewed in [Citation10,Citation11]).
Cytoplasmic trapping is a major problem for the effective intracellular delivery by the endocytic route. A number of solutions have been tested based on co-delivery of drugs or endosome-disruptor peptides, such as Ca2+ [Citation12], HIV Tat peptide [Citation10], chloroquine [Citation13], endosome-disruptive carriers (polyetylenimine [PEI], polyamidoamine [PAMAM]) [Citation14]. Studies here use photochemical internalization technology (PCI) [Citation15] to overcome the limitation of low cellular internalization efficiency [Citation16,Citation17], which is based on co-delivery of a membrane-soluble photosensitizer. Brief illumination generates short-lived singlet oxygen, which disrupts down the endosomal membrane with high efficiency and minimal effects on other cellular components.
Uptake across the nuclear envelope and recruitment to repair foci were achieved using an appropriately modified fragment of the human 53BP1 protein, which contains tandem Tudor domains that bind avidly to dimethyl histone residues that are exposed at repair sites [Citation18–21]. Although 53BP1 has been studied extensively, to our knowledge the present studies are the first to use this protein as part of a delivery platform for further investigation of DSB repair machinery or therapeutic purpose, and so on. Two 53BP1-based platforms were tested, one based on transferrin-53BP1 conjugate and the other on a PTD-53BP1 fusion.
Results indicate that platforms based on the combination of components and technologies discussed here were able to deliver the heterologous biotin-binding protein, streptavidin, to repair sites. Streptavidin has no intrinsic affinity for repair sites and thus serves as a model cargo in these proof-of-concept experiments. The approach is readily generalized for co-delivery of any material that can be biotinylated, including peptides, proteins, or other nanomaterials.
Materials and methods
Expression and characterization of a 53BP1 fragment
Construction, Escherichia coli expression, and purification of a fragment of the human 53BP1 gene encompassing residues 1053 to 1711 are detailed in Supplementary Material. The expressed fragment contained an N-terminal hexahistidine tag, a nuclear localization signal, and a biotin acceptor peptide tag. One version of the protein also contained an HIV Tat protein-derived PTD upstream of the biotin acceptor tag. Dimethyl histone binding experiments were performed using H4K20me2 peptide (RHRKme2VLRD, Abcam, Cambridge, MA in 10 μl of 10 mM Tris-HCl (pH 8.0), 150 mM KCl, 0.5 mM EDTA, 0.1% Triton-X 100, 12.5% Glycerol, 2 mM dithiothreitol. Complexes were analyzed (without heating) by non-reducing SDS-PAGE and co-migration of peptide with 53BP1 was determined by immunoblotting with anti-H4K20me2 antibody (1:2000, ab9052, Abcam, Cambridge, UK) and horseradish peroxidase-conjugated goat anti-rabbit IgG (1:10,000 Millipore, Burlington, MA) using ECL Western blotting substrate (Pierce) for visualization. Quantification was performed using Image J (rsbweb.nih.gov). Histone binding was also evaluated by pull-down assays. H4K20me1, H4K20me2 or H4K20me3 peptides (50 µg) were coupled to 200 µl DADPA UltraLink resin using carbodiimide chemistry (CarboxyLink Immobilization Kit, Thermo Scientific, Waltham, MA). Purified 53BP1 fragments (20 μg) and resin (15 μl) were incubated in a volume of 200 μl PBS at 4 °C. After 4 h, bound proteins were analyzed by SDS-PAGE. DNA binding was performed by incubating 53BP1 fragments with 2.5 nM HindIII-digested pBluescript DNA in 10 μl of 10 mM Tris-HCl (pH 8.0), 150 mM KCl, 0.5 mM EDTA, 0.1% Triton-X 100, 12.5% Glycerol, 2 mM dithiothreitol. After 30 min at room temperature, samples were analyzed by 0.6% agarose gel electrophoresis. 53BP1 biotinylation was determined by incubating expressed protein (20 µg) with 10 µl (packed volume) high capacity streptavidin-agarose (Thermo Scientific, Rockford, IL) in PBS at room temperature for 1 h in a volume of 200. Bound protein was analyzed by SDS-PAGE.
Assembly of the complete delivery system
Holo-transferrin (ProSpec, East Brunswick, NJ) (10 mg/ml) was incubated with 1 mM N-succinimidyl-3-(2-pyridyldithio) propionate (SPDP, ThermoScientific, Rockford IL) in 500 µl phosphate-buffered saline (PBS) for 1 h at room temperature. The mixture was dialyzed against PBS, 53BP1 protein fragment was added (1 mg/ml, final reaction volume 2 ml in PBS) and the mixture was incubated at 4 °C overnight. The transferrin-53BP1 binary complex was separated from reactants by Superdex 200 chromatography (GE Healthcare Life Sciences, Piscataway, NJ), and then incubated with equimolar streptavidin-conjugated Alexa Fluor 488 (Invitrogen) for 1 h at room temperature.
Delivery of fluorescent model cargo to repair foci
U2OS 2–6-3 cells [Citation22] were seeded in 35 mm glass-bottom plates (MatTek Corp., Ashland, MA) 16 h prior to use. Cells were transferred into medium containing 20 nM of the assembled delivery vehicle. After 4–6 h, they were transferred to fresh medium containing 10 µM Hoechst 33342 (Invitrogen). After 1 h, this was replaced with medium neither containing dyes nor proteins, and live cell imaging was performed in the WeatherStation environmental chamber of a Deltavision Microscope using a 60× objective. Filter sets were as follows: Hoechst, 387 nm excitation/440 nm emission; SA-AF488, 485 nm excitation/525 nm emission; FM4-64, 560 nm excitation/607 nm emission.
In experiments where photochemical internalization was used to promote endosome escape, cells were pre-treated with 10 µg/ml disulfonated aluminum phthalocyanin (AlPcS2a, Frontier Scientific, Logan, UT) for 16–18 h, washed three times, incubated with delivery vehicle as above, washed, and exposed for 90 s to a 660 nm light source (90 watt LED grow light, LEDwholesaler.com, Burlingame, CA). The light source was masked to create an 11 cm aperture, which was covered with a long-pass filter (650 nm cutoff, Newport Corp., Irvine, CA) and used at a 14 cm working distance. Cells were allowed to recover in medium containing Hoechst dye, and live cell imaging was performed as above, using a 650 nm excitation/684 nm emission filter set for AlPcS2a.
To observe foci formation, cells were electroporated with mCherry-53BP1 (23) prior to seeding on glass-bottom plates. All other steps were the same, except that cells received 2 Gy γ-radiation using a 137Cs source (Nordion, Ottawa, Canada) as the last step in the procedure and were allowed to recover for 25–30 min prior to live cell imaging. The 560 nm excitation/607 nm emission filter set was used for mCherry.
Results
Overall design of the multi-component delivery platforms
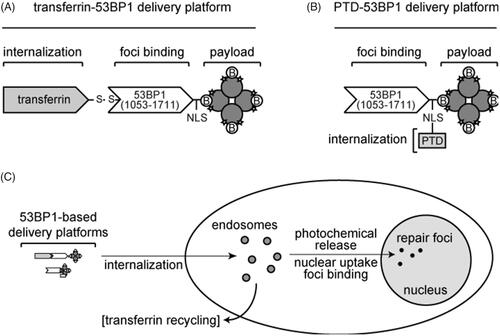
We developed two 53BP1-based platforms (. One is a multi-component system containing the iron carrier protein transferrin (), which is conjugated to an appropriately engineered 53BP1 protein by a scissile disulfide bond. The 53BP1 fragment has an N-terminal biotin acceptor peptide tag, which serves as a substrate for the endogenous Escherichia coli birA gene product. Use of the birA/biotin acceptor peptide system assures that each 53BP1 molecule incorporates only a single biotin residue, at a defined site that is well separated from the functional domains in the primary sequence. The 53BP1 complex is, in turn, coupled through the biotin residue to Alexa Fluor 488-streptavidin, which serves as the model cargo in these experiments. As depicted in the figure, 53BP1 occupies only one of the four biotin-binding sites in tetrameric streptavidin, with others available for potential attachment of additional ligands. The other delivery platform () incorporates a PTD fused to the primary amino acid sequence of 53BP1. The PTD provides internalization function in the absence of transferrin. The overall delivery strategy is summarized in , which depicts the ability of the 53BP1-based delivery platforms to undergo internalization, photochemical release, nuclear uptake, and foci binding. In the case of the transferrin-53BP1 platform, these processes are accompanied by recycling of the transferrin to the cell surface.
Figure 1. 53BP1-based delivery platforms. (A) Multicomponent transferrin-53BP1 delivery platform. The first component (transferrin) provides cell internalization function, the second (a 53BP1 fragment) provides a foci-binding function, and the third (streptavidin) is the model payload. Transferrin and 53BP1 are linked by a scissile disulfide bond. Other features include a nuclear localization sequence (NLS), biotin (B) molecules bound to the tetrameric streptavidin, and covalently linked Alexa Fluor 488 (stars). (B) PTD-53BP1 delivery platform. A protein transduction domain (PTD) is fused to the primary sequence of 53BP1 to provide cell internalization function. Other symbols are as in Panel A. (C) Overall scheme showing internalization, photochemical release, nuclear uptake, foci binding, and recycling of transferrin to the cell surface.
Expression and characterization of a recombinant 53BP1 fragment
The first step in assembling the delivery platforms was to create the 53BP1 fragments that form their central component. The minimal 53BP1 sequence that is required for binding to repair foci spans residues from 1231 to 1711 [Citation23]. It includes tandem Tudor domains, which directly contact dimethyl histone residues that are exposed in repair foci, and adjacent oligomerization motifs, which are required for high-affinity binding (). We expressed a slightly larger fragment (residues 1053–1711), based on preliminary experiments showing a much higher soluble yield in E. coli. To confirm that the expanded fragment was biologically active, we expressed it in mammalian cells, irradiated, and observed its localization to repair foci (Supplementary Figure 1). After validating the ability of the expanded fragment to migrate to repair foci, we next expressed and purified the fragment from E. coli. The E. coli expressed product contained a hexahistidine tag to facilitate purification, a eukaryotic nuclear localization sequence, and an acceptor peptide for E. coli BirA biotin ligase [Citation24], joined by a 27-residue linker to the 53BP1 sequences proper. We also prepared a second version of the 53BP1 fragment with an HIV Tat-derived protein-transduction domain (PTD), inserted immediately upstream of the biotin acceptor peptide tag.
Figure 2. Characterization of expressed 53BP1 (1053–1711) fragment. (A) Scale diagram of 53BP1. Expressed fragment contains tandem Tudor domains and oligomerization domains, but not C-terminal BRCT domains found in native 53BP1. (B) Co-electrophoresis assays to detect H4K20me2 peptide binding. Reactions contained 7.6 μM BSA, 1.0 μM 53BP1 fragment, or H4K20me2 peptide (4.2, 11.7, 23.4, or 46.8 μM). Immunoblotting with anti-H4K20me2 and Ponceau staining for total protein are shown, with ratios plotted (arbitrary units) in bottom panel. Dashed lines indicate background in the absence of peptide for each of the 53BP1 fragments. (C) Pull-down assays to detect H4K20me2 peptide binding. Indicated peptides were conjugated to agarose resin, incubated with 53BP1 fragments, and bound protein was analyzed by SDS-PAGE with Coomassie blue staining. Lanes labeled “on” contain 2 μg of input protein (equivalent to 10% of total input). Other lanes contain 30% of protein solubilized from each resin. Markers are indicated in kDa. (D) Streptavidin-agarose pull-down. SDS-PAGE analysis shows 10% of total input (T), 10% of free protein (F), and 10% of bound protein (B). Proteins were analyzed on a 4–12% Nu-PAGE SDS gel with Coomassie blue staining with markers indicated in kDa.
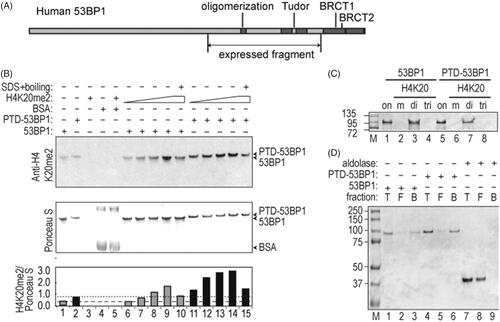
To evaluate the ability of purified 53BP1 fragments to bind to dimethyl histone residues in vitro, we performed a co-electrophoresis assay in which dimethyl H4K20 peptide was mixed with the purified 53BP1 fragments. The resulting complexes were resolved by partially denaturing SDS-PAGE, and the amount of bound H4K20me2 peptide was determined by immunoblotting (). Although anti-peptide antibody showed some cross-reactivity with 53BP1 in control reactions lacking peptide (lanes 1, 2), there was at least a four-fold increase in signal in the presence of increasing H4K20me2 peptide (lanes 6–9 and 11–14), and this signal was markedly reduced in samples denatured by heating in SDS prior to loading (lanes 10, 15). No H4K20me2 peptide signal was seen in control reactions containing bovine serum albumin in place of 53BP1 (lanes 4, 5). The specificity of binding was confirmed by comparing mono-, di-, and tri-methylated H4K20 peptides (). For this purpose, peptides were coupled to agarose support and used in a pull-down assay. The 53BP1 fragments bound to dimethyl H4K20-agarose (lanes 3, 7) but not to monomethyl H4K20-agarose or trimethyl H4K20-agarose (lanes 2, 4, 6, 8). Together, results indicate the formation of a stable, noncovalent 53BP1 complex that is specific for the dimethyl form of the H4K20 peptide.
In addition to dimethyl histone binding, the domain of 53BP1 that is responsible for binding to repair foci has been reported to have a weak, nonspecific DNA binding activity, which was confirmed in an electrophoretic mobility shift assay (Supplementary Figure 2).
Biotinylation was confirmed in streptavidin-agarose pull-down experiments using purified 53BP1 fragments (). 53BP1 was found in the total (T), and bound (B) fractions, but not in free (F) supernatant. Aldolase, a non-biotinylated protein control, was found in total protein and free supernatant fractions, but not in the bound fraction. Results indicate that biotinylation was achieved during E. coli expression, attributable to the BirA/biotin acceptor peptide system.
Assembly of the multi-component delivery platform
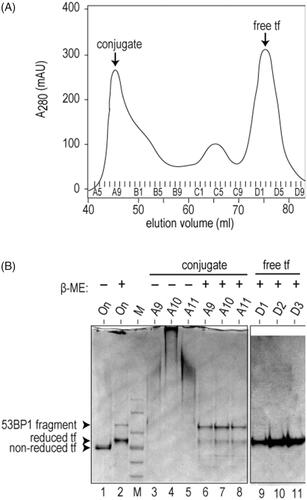
To assemble the transferrin-53BP1 delivery platform, holo-transferrin was first activated by controlled derivatization with the hetero-bifunctional cross-linker, SPDP. The resulting sulfhydryl-containing product was incubated with purified 53BP1 fragment under oxidizing conditions, and the reaction mixture was subjected to gel filtration chromatography. The first peak in the elution profile () represents disulfide-linked conjugate and the second major peak represents free transferrin. Consistent with these assignments, SDS-PAGE analysis of the first peak, under non-reducing conditions, revealed protein aggregates that do not enter the gel (, lanes 3–5), whereas the same analysis under reducing conditions regenerated the original transferrin and 53BP1 polypeptides (lanes 6–8). The purified transferrin-53BP1 conjugate, which bears a single site-specific biotin residue, was incubated with fluorescently labeled streptavidin to obtain the complete transferrin-53BP1 delivery platform. Similarly, to assemble the PTD-53BP1 delivery platform, the purified PTD-53BP1 fragment (also containing a single N-terminal biotin molecule) was incubated with fluorescently labeled streptavidin.
Figure 3. Assembly of transferrin-53BP1 delivery platform. (A) Gel filtration chromatogram of transferrin-53BP1 conjugation reaction. Absorbance at 280 nm (A280) is indicated in milliabsorbance units (mAU). Conjugate and free transferrin (tf) peaks are indicated. (B) SDS-PAGE analysis of fractions from panel A. Migration of 53BP1 fragment, reduced tf, and non-reduced tf are indicated. Loading buffer contained β-mercaptoethanol (β-ME) as indicated.
Internalization of 53BP1-based delivery platforms to human cells
The ability of the 53BP1-based delivery platforms to deliver streptavidin to the cell interior was characterized in U2OS 2–6-3 human osteosarcoma cells, a subclone of the U2OS osteosarcoma cell line that was used in previous studies of 53BP1 incorporation into repair foci [Citation23]. Cells were pre-incubated with 53BP1-containing complexes and a membrane-specific dye, FM4-64, for 4 to 6 h. They were allowed to recover for 1 h in the presence of the DNA binding dye, Hoechst 33342, and subjected to live cell imaging to follow the internalization of the complex. Results show uptake and punctate localization within the cytoplasm (. Streptavidin fluorescence was coincident with FM4-64 but did not overlap with the Hoechst 33342, indicating that the 53BP1-containing complexes were retained in cytoplasmic membranous structures, most likely endosomes, and did not enter the nucleus. A differential interference contrast image of treated cells showed prominent, refractile granules, which may correspond to the loaded endosomes (, top row, left panel). There was no uptake in the presence of free transferrin competitor, indicating that internalization occurs by a transferrin-saturable process (, second row)
Figure 4. Cell uptake of transferrin-53BP1 and PTD-53BP1 platforms. Columns show differential interference contrast (DIC) or staining with Hoechst 33342 (DNA), FM4-64 (endosomes) or Alexa Fluor 488 (53BP1-streptavidin). Rows are labeled to indicate which 53BP1-based delivery platform was tested. For the competition experiment with free transferrin, cells were preincubated for 30 min with medium containing 20 μM transferrin competitor, this was replaced with pre-warmed medium containing 5 μg/ml FM4-64 (Invitrogen) for 1 min, and this was replaced with medium containing Tf-53BP1 complex, FM4-64, and 20 μM transferrin. Control experiments (bottom two rows) were performed with FM4-64 and streptavidinAlexafluor 488 only (“no 53BP1”), or with 53BP1 fragment lacking both transferrin and PTD (“53BP1 (no tf, no PTD)”). Scale bars, 15 μm.
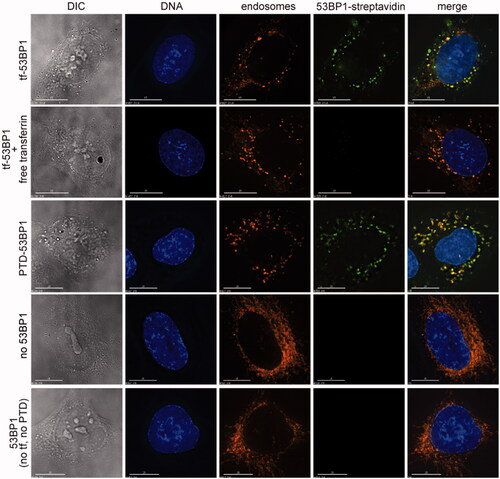
The PTD-53BP1 delivery platform behaved similarly to the transferrin-53BP1 platform. Streptavidin and FM4-64 fluorescence were again coincident, and streptavidin fluorescence was again excluded from the nuclei (, third row). In control experiments, there was no uptake of fluorescent streptavidin alone or of a 53BP1 complex lacking transferrin and PTD (, fourth and fifth rows). It was notable that in the control experiments, cytoplasmic granules were less prominent in the DIC images, and the FM4-64-labeled internal membrane had a more filamentous appearance. Together, results demonstrated the dependence of uptake on the inclusion of either transferrin or a PTD as part of the 53BP1-based delivery platform. They also highlight a major limitation of the system, which is retention in cytoplasmic vesicles.
Release of in vitro-labeled 53BP1 fragment from cytoplasmic vesicles
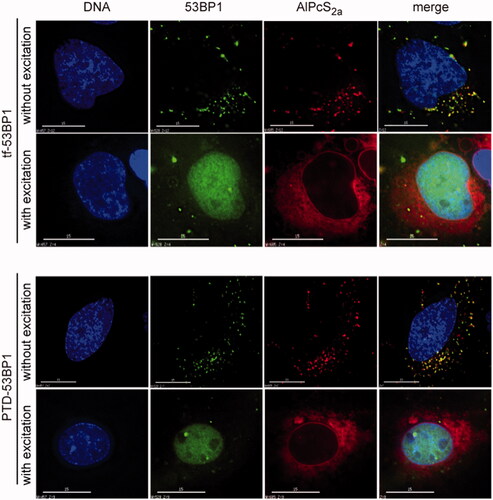
The cytoplasmic retention of the 53BP1-streptavidin complexes indicated the need for an additional step to promote the release and nuclear uptake. This was achieved by photochemical internalization using AlPcS2a, a lipid-soluble photosensitizer. In the absence of far-red light excitation, streptavidin fluorescence was seen as before in discrete, punctate structures (, “without excitation”). These punctate structures were coincident with AlPcS2a autofluorescent signal. Following brief excitation with >650 nm light, the pattern of localization was completely altered (, “with excitation”). The streptavidin-53BP1 complex migrated to the nucleoplasm, and the AlPcS2a assumed a more uniform cytoplasmic distribution, with some uptake into the nuclear envelope. The transferrin-53BP1 and PTD-53BP1 delivery platforms behaved very similarly. Together, these results indicate that photochemical internalization promoted synchronous and quantitative release from the cytoplasmic vesicles.
Figure 5. Photochemical release and nuclear uptake. Cells were incubated with indicated transferrin-53BP1 or PTD-53BP1 delivery platforms. Far-red light excitation was as indicated. AlPcS2a was detected based on its intrinsic fluorescence; other columns are as in .
Association with repair foci
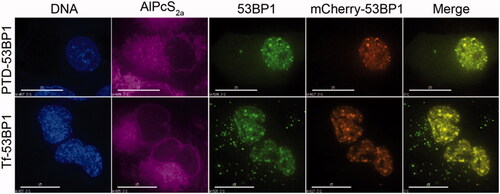
We next evaluated the ability of the 53BP1-based delivery platforms to direct association of fluorescent streptavidin with repair foci. Cells were transfected prior to plating with mCherry-53BP1 as an authentic marker of repair foci. After growth for 1 d to allow protein expression, they were incubated with 53BP1-based delivery platforms and AlPcS2a, then exposed briefly to far-red light. Immediately following these treatments, cells were exposed to 2 Gy of gamma radiation to induce DNA double-strand breaks and live-cell imaging was performed. Results show uptake of the 53BP1-containing complexes into discrete nuclear foci (. The focal pattern is in striking contrast to the homogeneous nucleoplasmic distribution seen in non-irradiated cells (compare and . The transferrin-53BP1 and PTD-53BP1 delivery platforms again showed a very similar ability to deliver fluorescent cargo to the target sites.
Figure 6. Localization of 53BP1-streptavidin complexes to repair foci. Cells were transfected with mCherry53BP1 expression plasmid(23) to establish a reference for foci formation, then with indicated transferrin-53BP1 or PTD-53BP1 delivery platforms. Cells were treated with 2 Gy of 137Cs radiation, and then live cell imaging was performed. Columns are labeled as in Figure 4, with the inclusion of a mCherry channel and merge channel which merged 53BP1-streptavidin and mCherry-53BP1 images. Note that AlPcS2a has been assigned a purple color to distinguish from mCherry. Scale bars, 15 μm.
Discussion
We describe here the development of an appropriately modified 53BP1 fragment that, in combination with other existing components and technologies, promotes localization of a fluorescent streptavidin model cargo, to sites of DNA repair. Successful delivery of the model cargo, which otherwise has no affinity for DNA repair sites, suggests that 53BP1-based delivery platforms could be generalized to deliver arbitrarily selected proteins, peptides, and other nanomaterials and nanomedicines to sites of DNA damage. Indeed, any material that can be biotinylated can potentially be co-delivered by this approach.
We tested two versions of the delivery platform, one using transferrin and the other using a PTD to effect cellular entry. Under the conditions used here, the two approaches gave equivalent results. The transferrin-53BP1 platform is particularly attractive for further development, as transferrin has been used to successfully deliver nanoparticles and other materials in vivo (reviewed in [Citation7]). There are at least two reports of successful use of transferrin receptor-targeted nanocarriers in clinical trials [Citation24,Citation25] and the approach is thus believed to face no fundamental barriers to clinical translation. PTDs effect cellular entry by a somewhat different process based on nonspecific charge-charge interactions with cell-surface components [Citation10,Citation11]). Unlike with transferrin, uptake is receptor-independent. PTD-mediated protein uptake has been widely applied in cultured cells, although the ability to apply the method in vivo, and in particular the prospects for clinical translation, are less certain.
As currently implemented, both 53BP1-based delivery platforms are dependent on photochemical internalization to relieve endosomal trapping. The AlPcS2a photosensitizer generates short-lived singlet oxygen, which disrupts membrane structure [Citation15]. PCI affords spatio-temporal control over the release of therapeutic agents and has been applied in nearly 100 cell lines and xenograft models and is also in clinical trials [Citation26]. The photochemical internalization effect operates on a ∼1 nm scale, and the reactive oxygen is not believed to reach the nucleus to induce genotoxicity. The PCI was chosen for endosomal disruption strategy for its high efficiency, and only highly efficient releasing could make visualization of foci forming by the streptavidin cargo. It’s reported almost complete (∼90%) endosomal release of DNA cargo through the PCI mechanism, and outperform commercial reagent 25k PEI by several hundred folds [Citation27]. Our result was similar to it. Whether it is possible to further develop the delivery platforms so that they are independent of PCI remains to be seen. There is at least one natural example of efficient trafficking of an extracellular receptor to the nucleus and indeed, to repair foci, which is epidermal growth factor receptor [Citation28]. We have also described a single chain antibody with a natural capability for endosome escape [Citation29]. Although the specific sequence element responsible for endosome escape in these settings are not yet fully defined, it is possible that it might eventually be used to engineer an endosome escape capability into the 53BP1-based delivery platforms.
The 53BP1-based delivery platforms have several potential applications. One is in the area of real-time imaging, as in the studies here where an organic fluorophore was linked to the streptavidin cargo. The use of a physical method of protein delivery, rather than a method based on gene expression, expands the palette of potential fluorophores to include synthetic materials such as organic dyes or quantum dots that are resistant to bleaching or that have other distinctive photophysical properties. In addition, the combination of endocytosis-mediated uptake and photochemical internalization allows a labeled species to be released synchronously and instantaneously within a cell population. This stands in contrast to methods based on the transfection of fluorescent reporter transgenes, where expression occurs asynchronously over a period of several days.
Other potential applications for 53BP1-based delivery platforms are in the areas of radiation therapy and gene correction. Radiation therapy kills tumor cells in proportion to the number of unrepaired DSBs. Delivery of macromolecular agents that interfere with repair foci assembly, such as a peptide inhibitor, could provide a potential means to sensitize target cells and reduce the radiation dose needed for tumor control. Gene correction is a method to treat genetic disease. It relies on targeted incision of the genome by an engineered nuclease, followed by homologous recombination repair using a donor template with the correct gene sequence. The efficiency of the process is restricted because homologous recombination operates in competition with nonhomologous end joining (NHEJ), which is the default pathway for DSB repair in mammalian cells. Delivery of a therapeutic agent that suppresses nonhomologous end joining in favor of productive homologous recombination repair pathway could provide a way to increase correction frequency to clinically useful levels. In both these examples, 53BP1-based platforms may afford a way to deliver peptides, aptamers, or other therapeutic agents at high local concentrations in the vicinity of the repair complex.
In the future, with 53BP1-based platforms described here, we are capable to visualize fluorescein or quantum dot labeled DSB repair proteins, or to deactivate some DSB repair components by introducing targeting inhibitors in NHEJ or HR pathway for detailed machinery investigation, or we can detour NHEJ pathway to HR pathway in vitro or in vivo by delivering suppressors targeting NHEJ key components to acquire high-fidelity DSB repair for gene correction therapy or gene editing research, or of course, we can also detour HR pathway by introducing suppressors targeting HR key components to obtain genomic instability for radiosensitivity increase in cancer cells or sensitivity increase for some targeted therapy medicine like PARP inhibitor. Besides these, as a functional and effective tool, 53BP1-based platforms could serve as a good delivery strategy for a wide range of field including research and clinical treatment.
Cao_Supplementary.docx
Download ()Acknowledgements
We thank Dr. David Spector and Dr. Ileng Kumaran (Cold Spring Harbor Laboratory, Cold Spring Harbor NY) for the gift of U2OS-2–6-3 cells, the members of the Nanomedicine Center for Nucleoprotein Machines for helpful discussions, and the Georgia Health Sciences University Imaging Core Facility for their services.
Additional information
Funding
Related Research Data
References
- Yoo S, Dynan WS. Characterization of the RNA binding properties of Ku protein. Biochemistry 1998;37:1336–1343.
- Kim CH, Park SJ, Lee SH. A targeted inhibition of DNA-dependent protein kinase sensitizes breast cancer cells following ionizing radiation. J Pharmacol Exp Ther. 2002;303:753–759.
- Li S, Takeda Y, Wragg S, et al. Modification of the ionizing radiation response in living cells by an scFv against the DNA-dependent protein kinase. Nucleic Acids Res. 2003;31:5848–5857.
- Salles B, Calsou P, Frit P, et al. The DNA repair complex DNA-PK, a pharmacological target in cancer chemotherapy and radiotherapy. Pathol Biol (Paris). 2006;54:185–193.
- Taylor KN, Eskander RN. PARP inhibitors in epithelial ovarian cancer. Recent Pat Anticancer Drug Discov. 2018;13:145–158.
- Dizdar O, Arslan C, Altundag K. Advances in PARP inhibitors for the treatment of breast cancer. Expert Opin Pharmacother. 2015;16:2751–2758.
- Qian ZM, Li H, Sun H, et al. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol Rev. 2002;54:561–587.
- Low PS, Kularatne SA. Folate-targeted therapeutic and imaging agents for cancer. Curr Opin Chem Biol. 2009;13:256–262.
- Atkinson SF, Bettinger T, Seymour LW, et al. Conjugation of folate via gelonin carbohydrate residues retains ribosomal-inactivating properties of the toxin and permits targeting to folate receptor positive cells. J Biol Chem. 2001;276:27930–27935.
- Chauhan A, Tikoo A, Kapur AK, et al. The taming of the cell penetrating domain of the HIV Tat: myths and realities. J Control Release. 2007;117:148–162.
- Murriel CL, Dowdy SF. Influence of protein transduction domains on intracellular delivery of macromolecules. Expert Opin Drug Deliv. 2006;3:739–746.
- Shiraishi T, Pankratova S, Nielsen PE. Calcium ions effectively enhance the effect of antisense peptide nucleic acids conjugated to cationic tat and oligoarginine peptides. Chem Biol. 2005;12:923–929.
- Dalluge R, Haberland A, Zaitsev S, et al. Characterization of structure and mechanism of transfection-active peptide-DNA complexes. Biochim Biophys Acta. 2002;1576:45–52.
- Berg K, Moan J. Lysosomes as photochemical targets. Int J Cancer. 1994;59:814–822.
- Berg K, Selbo PK, Prasmickaite L, et al. Photochemical internalization: a novel technology for delivery of macromolecules into cytosol. Cancer Res. 1999;59:1180–1183.
- Lee CS, Park W, Park SJ, et al. Endolysosomal environment-responsive photodynamic nanocarrier to enhance cytosolic drug delivery via photosensitizer-mediated membrane disruption. Biomaterials 2013;34:9227–9236.
- Kim KS, Park W, Na K. Gadolinium-chelate nanoparticle entrapped human mesenchymal stem cell via photochemical internalization for cancer diagnosis. Biomaterials 2015;36:90–97.
- Iwabuchi K, Basu BP, Kysela B, et al. Potential role for 53BP1 in DNA end-joining repair through direct interaction with DNA. J Biol Chem. 2003;278:36487–36495.
- Huyen Y, Zgheib O, Ditullio RA, Jr, et al. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 2004;432:406–411.
- Botuyan MV, Lee J, Ward IM, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 2006;127:1361–1373.
- Zgheib O, Pataky K, Brugger J, et al. An oligomerized 53BP1 tudor domain suffices for recognition of DNA double-strand breaks. Mol Cell Biol. 2009;29:1050–1058.
- Janicki SM, Tsukamoto T, Salghetti SE, et al. From silencing to gene expression: real-time analysis in single cells. Cell 2004;116:683–698.
- Cao Z, Kuhne WW, Steeb J, et al. Use of a microscope stage-mounted Nickel-63 microirradiator for real-time observation of the DNA double-strand break response. Nucleic Acids Res. 2010;38:e144.
- Weaver M, Laske DW. Transferrin receptor ligand-targeted toxin conjugate (Tf-CRM107) for therapy of malignant gliomas. J Neurooncol. 2003;65:3–13.
- Davis ME, Zuckerman JE, Choi CH, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010;464:1067–1070.
- Selbo PK, Weyergang A, Hogset A, et al. Photochemical internalization provides time- and space-controlled endolysosomal escape of therapeutic molecules. J Control Release. 2010;148:2–12.
- Xu X, Li Y, Liang Q, et al. Efficient gene delivery mediated by a helical polypeptide: Controlling the membrane activity via multivalency and light-assisted photochemical internalization (PCI). ACS Appl Mater Interfaces. 2018;10:256–266.
- Lin SY, Makino K, Xia W, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–808.
- Xiong H, Lee RJ, Haura EB, et al. Intranuclear delivery of a novel antibody-derived radiosensitizer targeting the DNA-dependent protein kinase catalytic subunit. Int J Radiat Oncol Biol Phys. 2012;83:1023–1030.