
Abstract
Acute lung injury (ALI) is a serious threat for health and lives worldwide. Recently, lncRNA MEG3 has been well studied to participate in lung cancer, making us speculate that it may be essential for ALI. ALI was simulated by treatment of LPS in human lung fibroblast WI-38 cells and human PMVECs. Cell viability, migration, apoptosis and MEG3 level were assessed by CCK-8, Transwell, flow cytometry/Western blot and qRT-PCR assays, respectively. Utilizing bioinformatics methods, luciferase activity assay and assessments of cell viability, migration and apoptosis, the possible interacted microRNA were explored. We found that, LPS induced decreases of cell viability, migration and MEG3 expression but promoted cell apoptosis. MEG3 knockdown aggravated LPS-induced injury of WI-38 cells and PMVECs. MEG3 acted as a sponge of miR-4262 and effects of MEG3 knockdown could be alleviated by miR-4262 inhibition. Krüppel-like factor 4 (KLF4) was negatively regulated by miR-4262, and its overexpression ameliorated LPS-induced cell injury. LPS-induced alterations of key kinases in the PI3K/AKT and JAK/STAT pathways were reversed by KLF4 overexpression. In conclusion, MEG3 was down-regulated by LPS and its knockdown aggravated LPS-induced injury of human lung cells through miR-4262-mediated down-regulation of KLF4. The PI3K/AKT and JAK/STAT pathways were implicated.
Introduction
Acute lung injury (ALI) is a clinical severe respiratory disorder and often leads to acute respiratory distress syndrome, resulting in remarkable annual morbidity, mortality and healthcare expenditures [Citation1,Citation2]. ALI is a multifactorial illness, which may be induced by diverse aetiologies, such as direct injury to the lung including pneumonia, toxic inhalation, near-drowning and aspiration, and indirect causes including burns, massive blood transfusion, sepsis, etc. [Citation3]. Even though extensive literatures have focused on the relevant pathogenesis of ALI, the treatment of ALI remains to be a task with tremendous hardship [Citation4]. Thus, innovative therapeutic strategies are of greatly unmet needs.
Lipopolysaccharide (LPS) is an endotoxin expressed on the outer membrane of Gram-negative bacteria [Citation5]. Currently, LPS is accepted to induce release of pro-inflammatory cytokines, resulting in acute tissue injury [Citation3,Citation6]. Intratracheal administration or intranasal instillation of LPS has been widely used for the construction of animal model with ALI [Citation7,Citation8]. LPS-induced lung injury in the mice is a robust experimental model for the studies of ALI [Citation9]. A previous study has revealed the possible roles of hedgehog signaling pathway in the LPS-induced cell injury of pulmonary microvascular endothelial cells (PMVECs) [Citation10]. Thus, cell model with LPS-induced cell injury is of great importance for the deep mechanism research of ALI.
Long non-coding RNAs (LncRNAs) are a loosely classified group of long RNA transcripts (longer than 200 nucleotides) which were involved in diverse biological processes, including proliferation, migration and apoptosis [Citation11,Citation12]. With the developments of epigenomic technologies and bioinformatics, the number of novel LncRNAs is climbing [Citation13]. Currently, several LncRNAs are reported to be involved in lung diseases, such as NANCI [Citation14] and GAS5 [Citation15]. LncRNA maternally expressed gene 3 (MEG3), transcribed from chromosome 14q32, is aberrantly expressed in multiple cancers [Citation16] and its down-regulation has been reported to augment cisplatin resistance of lung cancer [Citation17]. However, very little is known about the functional roles of MEG3 in lung injury.
Migration and proliferation of lung fibroblasts were observed at the early stage of ALI [Citation18]. Additionally, lung fibroblasts are described as an essential player of lung repair during ALI [Citation19]. Herein, in our study, human lung normal fibroblast WI-38 cells were treated with LPS to simulate ALI. Then, the expression and the roles of MEG3 in the LPS-treated WI-38 cells were explored. In the meantime, the effects of MEG3 on LPS-treated human PMVECs were also studied to support the results from WI-38 cells. Moreover, the molecular mechanism and possibly involved signaling pathways in the MEG3-associated modulation were investigated.
Materials and methods
Cell culture and treatment
WI-38 cells (American Type Culture Collection, Manassas, VA) were cultured in Dulbecco’s modified Eagle medium (DMEM; GIBCO, Grand Island, NY) containing 10% foetal bovine serum (FBS; Invitrogen, Carlsbad, CA), antibiotic/antimycotic solution (100 units) and gentamycin (50 µg/mL) with Na pyruvate (1 mM). Human PMVECs (Biofavor Biotech Service Co., Ltd., Wuhan, China) were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA) containing 10% FBS, 1% penicillin/streptomycin and 0.5% fungizone (Invitrogen, Carlsbad, CA). Cells were grown at 37 °C in a humidified atmosphere (5% CO2 in air). To construct cell model with cell injury, cells were stimulated with E. coli-derived LPS (10 μg/mL; Sigma-Aldrich, St. Louis, MO) for 5 h [Citation20].
Cell counting kit-8 (CCK-8) assay
Cell viability was measured by a CCK-8 assay. Briefly, cells were seeded into a 96-well plate at a density of 5 × 103 cells/well. After treatments, 10 μL CCK-8 solution (Dojindo Molecular Technologies, Gaithersburg, MD) was added to the culture medium of each well, followed by 1 h of incubation at 37 °C in a humidified incubator containing 5% CO2 in air. Then, a Microplate Reader (Bio-Rad, Hercules, CA) was used for measurement of absorbance at 450 nm. Cell viability was presented as the absorbance in relation to the control group (set as 100%).
Migration assay
Cell migration was determined using Transwell chambers with 8-μm sized pores (BD Biosciences, Bedford, MA). Briefly, treated-cells were resuspended in FBS-free medium and seeded onto the upper compartments with a volume of 200 μL. Then, 600 μL complete medium was added into the lower compartments. Following 24 h of incubation at 37 °C, cells were fixed by methanol. Afterwards, the non-traversed cells staying on the upper surface of the filter were removed carefully with a cotton swab, whereas the cells that traversed to the lower side of the filter were stained with crystal violet and were counted under an inverted microscope (Olympus Optical Co., Ltd., Tokyo, Japan).
Apoptosis assay
Cell apoptosis was estimated by propidium iodide (PI) and fluorescein isothiocyanate (FITC)-conjugated Annexin V staining. In brief, treated-cells were washed by phosphate-buffered saline (PBS), fixed in 70% ethanol and washed again by PBS. Then, cells were collected and resuspended in binding buffer from an Annexin V-FITC/PI apoptosis detection kit (Beijing Biosea Biotechnology, Beijing, China). Cells were subsequently stained by Annexin V-FITC and PI following the information of the manufacture. The apoptotic cells were differentiated by using a flow cytometer (Beckman Coulter, Fullerton, CA). Percentage of apoptotic cells was analysed by FlowJo software (Tree Star, San Carlos, CA).
Quantitative reverse transcription PCR (qRT-PCR)
For the measurement of MEG3 and mRNA, total RNA was extracted from treated-cells using Trizol reagent (Invitrogen, Carlsbad, CA) following the manufacturer’s recommendation. Then, quantitation of MEG3 was performed with One Step SYBR® PrimeScript™ PLUS RT-RNA PCR Kit (TaKaRa Biotechnology, Dalian, China). Quantitation of mRNAs was performed with MultiScribe™ Reverse Transcriptase kit (Applied Biosystems, Foster City, CA) and SYBR™ Green PCR Master Mix (Applied Biosystems, Foster City, CA) for reverse transcription and real-time PCR, respectively, according to the protocols provided by the suppliers. For the measurement of miR-4262, small RNA was isolated from treated-cells using RNAiso for Small RNA (TaKaRa Biotechnology, Dalian, China) following the protocol of supplier. Then, the Taqman MicroRNA Reverse Transcription Kit and Taqman Universal Master Mix II (both from Applied Biosystems, Foster City, CA) were used for reverse transcription and real-time PCR, respectively, on the basis of the information of supplier. Primers were all designed and synthesized by Sangon Co., Ltd. (Shanghai, China). Relative expression was calculated according to the 2–ΔΔCt method described previously [Citation21], normalizing to U6 (for miR-4262) or GAPDH (for MEG3 and mRNAs).
Generation of stably transfected cells and miRNA transfection
Short-hairpin RNA directed against human MEG3 or Krüppel-like factor (KLF) 4 was sub-cloned into the pGPU6/Neo plasmid (GenePharma, Shanghai, China) to generate sh-MEG3 or sh-KLF4. The plasmid carrying a non-targeting sequence was used as a negative control (NC) of sh-MEG3 or sh-KLF4, termed sh-NC. For overexpression of KLF4, the full-length KLF4 sequences were ligated into pEX-2 plasmid (GenePharma, Shanghai, China) to generate pEX-KLF4, control of which was the empty pEX-2 plasmid, termed pEX. The Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA) was utilized for cell transfection in line with the manufacturer’s instructions. G418 (0.5 mg/mL; Sigma-Aldrich, St. Louis, MO) was added to the culture medium for the selection of stably transfected cells. G418-resistant cell clones were established after approximately 4 weeks. MiR-4262 mimic, scramble miRNAs, miR-4262 inhibitor and its NC were purchased from Life Technologies Corporation (Gaithersburg, MD). Cell transfection with miRNAs was also performed with Lipofectamine 3000 reagent, and cells were harvested at 72 h post-transfection in the subsequent experiments.
Construction of reporter vectors and luciferase reporter assay
The fragment from human MEG3 containing the putative miR-4262 binding site was sub-cloned into a pmirGLO plasmid (Promega, Madison, WI) to form the reporter vector MEG3-wild-type (MEG3-Wt). To mutate the putative binding site of miR-4262 in the MEG3, the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) was utilized following the manufacturer’s suggestions to generate MEG3-mutated-type (MEG3-Mt). After sequencing, 293T cells were co-transfected with miR-4262 mimics (scramble miRNAs) and MEG3-Wt (MEG3-Mt), followed by the determination of luciferase activity using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI).
Western blot analysis
The proteins of treated-cells were extracted using RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) supplemented with protease inhibitors (Applygen Technologies Inc., Beijing, China). After quantification with the BCA™ Protein Assay Kit (Pierce, Appleton, WI), equivalent proteins were separated by SDS-PAGE. Then, proteins in the gels were blotted to polyvinylidene difluoride (PVDF) membranes, followed by blockage with 5% bovine serum albumin (BSA). PVDF membranes were then incubated at 4 °C overnight with primary antibodies against B cell lymphoma-2 (Bcl-2, ab32124), Bcl-2-associated X protein (Bax, ab182733), pro caspase-3 (ab90437), cleaved caspase-3 (ab2302), pro caspase-9 (ab32539), cleaved caspase-9 (ab2324), KLF4 (ab72543), phosphatidylinositol-3-kinase (PI3K, ab86714), phospho-PI3K (p-PI3K, ab182651), Janus-activated kinase-1 (JAK1, ab133666), phospho-JAK1 (p-JAK1, ab138005), signal transducer and activator of transcription-1 (STAT1, ab3987), phospho-STAT1 (p-STAT1, ab109461), STAT3 (ab68153), phospho-STAT3 (p-STAT3, ab76315), GAPDH (ab128915) (all from Abcam, Cambridge, UK), phospho-AKT (p-AKT, #4060) and AKT (#4685) (both from Cell Signaling Technology, Beverly, MA). After rinsing and incubation with secondary antibodies marked by horseradish peroxidase for 1 h at room temperature, membranes were transferred into the Bio-Rad ChemiDoc™ XRS system for the visualization of the bands with chemiluminescence (ECL) system (Amersham Biosciences, Piscataway, NJ). The ImageJ software (National Institutes of Health, Bethesda, MA) was used to detect the intensity of the bands.
Statistical analysis
All experiments were repeated three times. The results were presented as the mean ± standard error of the mean (SEM). Statistical analysis was performed using Graphpad Prism 5 software (GraphPad, San Diego, CA). The p values were calculated using the one-way analysis of variance (ANOVA). A p < .05 was considered as a significant difference.
Results
LPS induced WI-38 cell injury
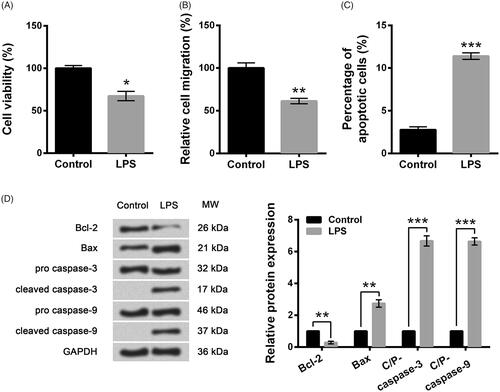
LPS-induced cell injury was evaluated by the alterations of cell viability, migration, apoptosis as well as apoptosis-associated proteins. Compared with control group, LPS significantly reduced cell viability (p < .05, ), migration (p < .01, ) and markedly enhanced the percentage of apoptotic cells (p < .001, ). shows protein expression of Bcl-2 was markedly down-regulated (p < .01) but expression of Bax, cleaved/pro caspase-3 and cleaved/pro caspase-9 was remarkably up-regulated (p < .01 or p < .001) after stimulation of LPS. All the results demonstrated that cell model with cell injury was successfully constructed after stimulation of LPS for 5 h.
Figure 1. LPS induced WI-38 cell injury. (A) Cell viability by CCK-8 assay. (B) Cell migration by Transwell assay. (C) Cell apoptosis by flow cytometry. (D) Expression of apoptosis-associated proteins by Western blot analysis. Data presented are the mean ± SEM of at least three independent experiments. *p < .05; **p < .01; ***p < .001. C/P-: cleaved/pro; MW: molecular weight.
MEG3 was down-regulated by LPS in WI-38 cells and PMVECs
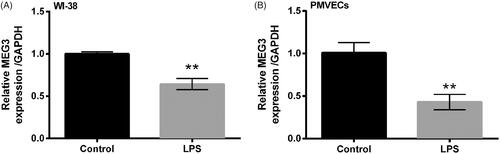
The expression of MEG3 was assessed by qRT-PCR after treatment of LPS. Compared with the control group, MEG3 was significantly down-regulated by LPS in both WI-38 cells (p < .01, ) and PMVECs (p < .01, ), indicating the possible involvements of MEG3 in the LPS-induced injury of lung cells.
Figure 2. Long non-coding RNA MEG3 was down-regulated by LPS treatment. Expression of MEG3 in WI-38 cells (A) and human PMVECs (B) was assessed by qRT-PCR. Data presented are the mean ± SEM of at least three independent experiments. **p < .01.
Knockdown of MEG3 aggravated LPS-induced injury of lung cells
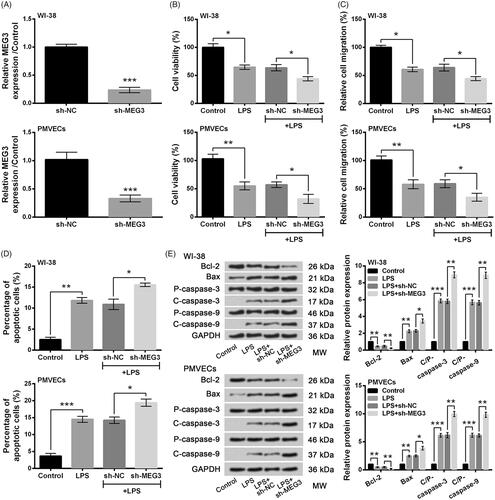
To explore the effects of MEG3 on LPS-induced cell injury, MEG3 was significantly down-regulated in WI-38 cells and PMVECs by transfection with sh-MEG3 compared to the sh-NC group (both p < .001, ). When compared to the LPS + sh-NC group, LPS-induced decreases of cell viability and migration were markedly decreased by MEG3 knockdown (all p < .05, ), whereas LPS-induced increase of apoptotic cells was significantly increased by MEG3 knockdown (both p < .05, ) in WI-38 cells and PMVECs. Meanwhile, LPS-induced alterations of Bax, Bcl-2, cleaved/pro caspase-3 and cleaved/pro caspase-9 were further augmented by MEG3 knockdown compared with the LPS + sh-NC group (p < .05 or p < .01, ). Results above suggested that LPS-induced injury of lung cells could be aggravated by MEG3 knockdown.
Figure 3. Knockdown of long non-coding RNA MEG3 aggravated LPS-induced injury of lung cells. (A) Expression of MEG3 by qRT-PCR. (B) Cell viability by CCK-8 assay. (C) Cell migration by Transwell assay. (D) Cell apoptosis by flow cytometry. (E) Expression of apoptosis-associated proteins by Western blot analysis. Data presented are the mean ± SEM of at least three independent experiments. *p < .05; **p < .01; ***p < .001. P-: pro; C-: cleaved; C/P: cleaved/pro; MW: molecular weight.
MiR-4262 was negatively correlated with MEG3 expression
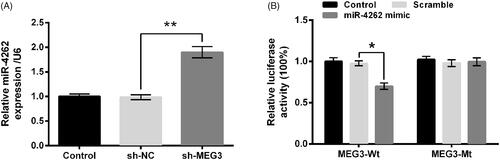
Owing to the development of bioinformatics, MEG3 was predicted to bind to miR-4262. In , MEG3 knockdown dramatically up-regulated level of miR-4262 compared with the sh-NC group (p < .01), indicating that miR-4262 was negatively regulated by MEG3. In addition, the luciferase activity was markedly reduced by co-transfection with MEG3-Wt and miR-4262 mimic compared with co-transfection with MEG3-Wt and scramble miRNAs (p < .05), whereas the difference of luciferase between co-transfections with MEG3-Mt was non-significant, suggesting the direct binding of MEG3 to miR-4262 (). Thus, we concluded that miR-4262 was negatively correlated with MEG3 expression.
Figure 4. MiR-4262 was negatively correlated with long non-coding RNA MEG3 expression. (A) Expression of MEG3 by qRT-PCR. (B) Luciferase activity by luciferase reporter assay. Data presented are the mean ± SEM of at least three independent experiments. *p < .05; **p < .01.
Knockdown of MEG3 exacerbated LPS-induced WI-38 cell injury through up-regulating miR-4262
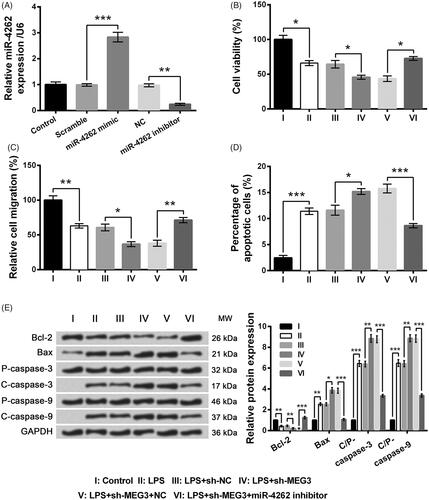
To explore the effects of miR-4262 on LPS-induced cell injury, WI-38 cells were transfected with miR-4262 mimic, miR-4262 inhibitor or their respective controls. In , miR-4262 was significantly up-regulated by transfection with miR-4262 mimic compared with the scramble group (p < .001) but was markedly down-regulated by transfection with miR-4262 inhibitor compared with the NC group (p < .01). Subsequent experiments showed MEG3 knockdown-induced alterations of cell viability, migration and apoptosis in LPS-treated WI-38 cells were observably reversed by miR-4262 inhibition compared with the LPS + sh-MEG3 + NC group (p < .05, cell viability in ; p < .01, migration in ; p < .001, apoptosis in ). Collectively, MEG3 knockdown might affect LPS-induced WI-38 cell injury through up-regulation of miR-4262.
Figure 5. Effects of long non-coding RNA MEG3 silence on LPS-induced WI-38 cell injury were alleviated by miR-4262 inhibition. (A) Expression of miR-4262 by qRT-PCR. (B) Cell viability by CCK-8 assay. (C) Cell migration by Transwell assay. (D) Cell apoptosis by flow cytometry. (E) Expression of apoptosis-associated proteins by Western blot analysis. Data presented are the mean ± SEM of at least three independent experiments. *p < .05; **p < .01; ***p < .001. P-: pro; C-: cleaved; C/P: cleaved/pro; MW: molecular weight; NC: negative control of miR-4262 inhibitor.
KLF4 was negatively modulated by miR-4262 expression
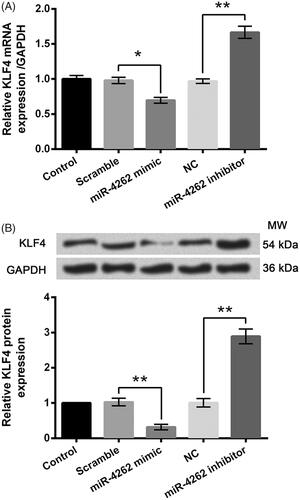
Utilizing bioinformatics methods, the possible interacted genes of miR-4262 were screened. Among those genes, KLF4 was selected to be fully investigated. In , mRNA and protein expression of KLF4 was down-regulated by miR-4262 overexpression (p < .05 or p < .01) while were up-regulated by miR-4262 inhibition (both p < .01), indicating the negative correlation between miR-4262 and KLF4.
Figure 6. MiR-4262 negatively regulated the expression of KLF4 in WI-38 cells. Expression levels of mRNA (A) and protein (B) were assessed by qRT-PCR and Western blot analysis, respectively. Data presented are the mean ± SEM of at least three independent experiments. *p < .05; **p < .01. NC: negative control of miR-4262 inhibitor; MW: molecular weight.
Aberrantly expressed KLF4 affected LPS-induced WI-38 cell injury
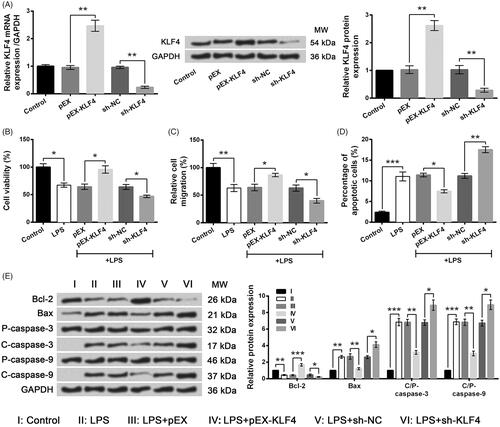
Next, different plasmids were respectively transfected into WI-38 cells and the stable transfected cells were selected. In , mRNA and protein levels of KLF4 were markedly increased by pEX-KLF4 (both p < .01) but were remarkably decreased by sh-KLF4 (both p < .01). Following experiments showed LPS-induced alterations of cell viability (), migration () and apoptosis () were significantly exacerbated by KLF4 silence (p < .05 or p < .01) while were markedly ameliorated by KLF4 overexpression (p < .05, p < .01 or p < .001). Results suggested that aberrantly expressed KLF4 affected LPS-induced WI-38 cell injury.
Figure 7. LPS-induced WI-38 cell injury was ameliorated by KLF4 overexpression but was aggravated by KLF4 knockdown. (A) Expression of KLF4 by qRT-PCR and Western blot analysis. (B) Cell viability by CCK-8 assay. (C) Cell migration by Transwell assay. (D) Cell apoptosis by flow cytometry. (E) Expression of apoptosis-associated proteins by Western blot analysis. Data presented are the mean ± SEM of at least three independent experiments. *p < .05; **p < .01; ***p < .001. P-: pro; C-: cleaved; C/P: cleaved/pro; MW: molecular weight.
KLF4 alleviated the LPS-induced inhibition of the PI3K/AKT and JAK/STAT pathways
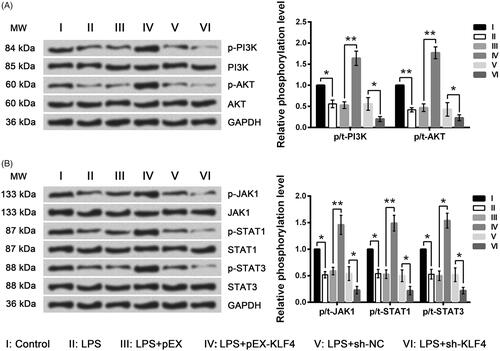
To reveal the underlying mechanisms of MEG3-associated regulations, the phosphorylated levels of key kinases in the PI3K/AKT and JAK/STAT pathways were assessed by Western blot analysis. Results in showed phosphorylated levels of PI3K, AKT, JAK1, STAT1 and STAT3 were all reduced by LPS (p < .05 or p < .01). Moreover, the LPS-induced down-regulations of phosphorylated kinases were further down-regulated by KLF4 silence (all p < .05) but were reversed by KLF4 overexpression (p < .05 or p < .01). Collectively, the LPS-induced inhibition of the PI3K/AKT and JAK/STAT pathways could be reversed by KLF4.
Figure 8. LPS-induced inhibition of the PI3K/AKT and JAK/STAT signaling pathways was further inhibited by KLF4 knockdown but was reversed by KLF4 overexpression. Expression of key kinases involved in the PI3K/AKT (A) and JAK/STAT (B) signaling pathways was assessed by Western blot analysis. MW: molecular weight.
Discussion
With the high incidence and mortality, ALI is a critical illness that threats health and lives worldwide [Citation22]. In our study, MEG3 was identified to be down-regulated in LPS-treated WI-38 cells and human PMVECs. LPS-induced cell injury, presenting as the decreases of cell viability and migration and the increase of cell apoptosis, was aggravated by MEG3 knockdown by up-regulating miR-4262. KLF4, negatively regulated by miR-4262, could ameliorate LPS-induced WI-38 cell injury through activating the PI3K/AKT and JAK/STAT signaling pathways.
During ALI, large amount of neutrophils, inflammatory mediators, proteases and oxidants were accumulated, resulting in vascular endothelia and alveolar epithelial cell damage or death. Then, the blood-alveolar barrier is disrupted, leading to intrapulmonary pulmonary oedema and impaired gas exchange [Citation23]. In our study, the LPS-induced cell injury was evaluated according to the alterations of cell viability, migration and apoptosis. After stimulation with LPS, the cell viability and migration were reduced while cell apoptosis was elevated. Additionally, the pro-apoptotic Bax was up-regulated but anti-apoptotic Bcl-2 was down-regulated after LPS treatments, elucidating that LPS could induce cell apoptosis through the mitochondrial-related pathway. In the meantime, the active caspase-9 and active caspase-3 were both increased by LPS treatments, which implied that LPS activated the caspase pathway. These two activated pathways induced by LPS might provide an explanation for the changed cell viability and apoptosis.
MEG3 has been widely explored in diverse cancer types including lung cancer [Citation24], thus we hypothesized that MEG3 might be of importance in ALI. First of all, the down-regulation of MEG3 in LPS-treated WI-38 cells and PMVECs powerfully suggested the possible association of MEG3 in LPS-induced lung cell injury. Then, MEG3 was stably knocked down, followed by assessments of cell viability, migration and apoptosis. Results presented that LPS-induced injury of WI-38 and PMVECs was aggravated by MEG3 knockdown. The inhibitory effect on cell proliferation as well as inducible effect on cell apoptosis of MEG3 knockdown in LPS-treated WI-38 cells differs from that in lung cancer reported in a previous literature [Citation25]. However, MEG3 knockdown has been stated to inhibit epithelial–mesenchymal transition in lung cancer [Citation26], which was consistent with the results in our study. The similar but not identical effects of MEG3 might be associated with the alterations from normal to cancer cells.
A previous study once stated that MEG3 repressed proliferation of hepatocellular carcinoma cells via negative modulation of miR-664 [Citation27]. To explore the molecular mechanism of the MEG3-associated modulations in LPS-treated lung cells, bioinformatics software was applied to analyse the possible relevant miRNAs of MEG3. In our study, miR-4262 was negatively correlated with MEG3 expression, and the subsequent luciferase assay also proved the direct binding between MEG3 and miR-4262. In addition, the effects of MEG3 knockdown on LPS-induced WI-38 cell injury were ameliorated by miR-4262 inhibition, illustrating that MEG3 knockdown functioned through up-regulating miR-4262.
MiRs are small non-coding RNAs that participate in physiology and disease through down-regulating target mRNAs [Citation28]. The roles of miR-4262 in cell proliferation, migration and apoptosis are controversial due to the different target mRNAs [Citation29,Citation30]. Thus, we next screened the possible interacted genes of miR-4262 via bioinformatics methods. KLF4 is a conserved transcriptional factor that participates in multiple cellular processes, such as cell proliferation, migration and apoptosis [Citation31]. KLF4 acts as either a tumor suppressor or an oncogene in diverse cancers, and the suppressive role of KLF4 in lung cancer has been proven [Citation32]. In another literature, ALI could be ameliorated by miR-7 deficiency through up-regulating KLF4 [Citation33]. Hence, in our study, we focused on the interaction between miR-4262 and KLF4. Results implied KLF4 was negatively correlated with miR-4262 level and KLF4 overexpression could alleviate LPS-induced WI-38 cell injury. The protective role of KLF4 overexpression in LPS-treated WI-38 cells was consistent with the literature described above [Citation33]. On the basis of the above results, it can be suggested that MEG3 knockdown might aggravate LPS-induced WI-38 cell injury through miR-4262-mediated down-regulation of KLF4.
Activation of the PI3K/AKT pathway has been proven to protect A549 cells from LPS-induced cell apoptosis [Citation34]. Insulin has been proven to alleviate LPS-induced ALI through activation of the PI3K/AKT pathway [Citation35]. Accumulating evidence has proven the JAK/STAT signaling pathway plays crucial roles in ALI [Citation36,Citation37]. In our study, these two signaling pathways were both inhibited by LPS, and were further inhibited by KLF4 knockdown. However, KLF4 overexpression could ameliorate the LPS-induced inhibition, indicating the possible involvements of the PI3K/AKT and JAK/STAT signaling pathways in the LPS-induced WI-38 cell injury. More details about the interactions between KLF4 and these two pathways should be investigated in the future.
To summarize, MEG3 was down-regulated by LPS and its down-regulation could exacerbate LPS-induced injury of lung cells through miR-4262-mediated silence of KLF4, involving the PI3K/AKT and JAK/STAT signaling pathways. This study suggested that MEG3 might be a potential volunteer for the treatment of ALI. However, this application needs more validations performed in vivo.
Disclosure statement
The authors declare that there is no conflict of interest.
References
- Altemeier WA, Chi FH, Matute-Bello G. Mouse models of acute lung injury. Berlin: Springer International Publishing; 2017.
- Jiang W, Luo F, Lu Q. The protective effect of Trillin LPS-induced acute lung injury by the regulations of inflammation and oxidative state. Chem-Biol Interact. 2016;243:127.
- Zhu GF, Guo HJ, Huang Y, et al. Eriodictyol, a plant flavonoid, attenuates LPS-induced acute lung injury through its antioxidative and anti-inflammatory activity. Exp Ther Med. 2015;10:2259.
- Chen T, Mou Y, Tan J, et al. The protective effect of CDDO-Me on lipopolysaccharide-induced acute lung injury in mice. Int Immunopharmacol. 2015;25:55–64.
- Maldonado RF, Sá-Correia I, Valvano MA. Lipopolysaccharide modification in Gram-negative bacteria during chronic infection. FEMS Microbiol Rev. 2016;40:480.
- Al-Harbi NO, Imam F, Al-Harbi MM, et al. Dexamethasone attenuates LPS-induced acute lung injury through inhibition of NF-κB, COX-2, and pro-inflammatory mediators. Immunol Investig. 2016;45:349.
- Ogata-Suetsugu S, Yanagihara T, Hamada N, et al. Amphiregulin suppresses epithelial cell apoptosis in lipopolysaccharide-induced lung injury in mice. Biochem Biophys Res Commun. 2017;484:422–428.
- Liu YL, Liu YJ, Liu Y, et al. Hydroxysafflor yellow A ameliorates lipopolysaccharide-induced acute lung injury in mice via modulating toll-like receptor 4 signaling pathways. Int Immunopharmacol. 2014;23:649–657.
- Wang R, Lu B, Gerard C, et al. C5L2, the second C5a anaphylatoxin receptor, suppresses LPS-induced acute lung injury. Am J Respir Cell Mol Biol. 2016;55:657.
- Yang Y, Li Q, Deng Z, et al. Protection from lipopolysaccharide-induced pulmonary microvascular endothelial cell injury by activation of Hedgehog signaling pathway. Mol Biol Rep. 2011;38:3615–3622.
- Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789.
- Zhang HM, Yang FQ, Chen SJ, et al. Upregulation of long non-coding RNA MALAT1 correlates with tumor progression and poor prognosis in clear cell renal cell carcinoma. Tumour Biol. 2015;36:2947.
- Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17:47–62.
- Zhang Y, Cheng HP, Bao TP, et al. Expression of long non-coding RNA NANCI in lung tissues of neonatal mice with hyperoxia-induced lung injury and its regulatory effect on NKX2.1. Zhongguo Dang Dai er ke za Zhi = Chin J Contemp Pediatr. 2017;19:215–221.
- Shi X, Sun M, Liu H, et al. A critical role for the long non-coding RNA GAS5 in proliferation and apoptosis in non-small-cell lung cancer. Mol Carcinog. 2015;54 (Suppl. 1):E1.
- Guo W, Dong Z, Liu S, et al. Promoter hypermethylation-mediated downregulation of miR-770 and its host gene MEG3, a long non-coding RNA, in the development of gastric cardia adenocarcinoma. Mol Carcinog. 2017;58:1924–1934.
- Xia Y, He Z, Liu B, et al. Downregulation of Meg3 enhances cisplatin resistance of lung cancer cells through activation of the WNT/β-catenin signaling pathway. Mol Med Rep. 2015;12:4530.
- Quesnel C, Nardelli L, Piednoir P, et al. Alveolar fibroblasts in acute lung injury: biological behaviour and clinical relevance. Eur Respir J. 2010;35:1312–1321.
- Villar J, Cabrera-Benitez NE, Ramos-Nuez A, et al. Early activation of pro-fibrotic WNT5A in sepsis-induced acute lung injury. Crit Care. 2014;18:568.
- Zhu Z, Dai J, Liao Y, et al. Sox9 protects against human lung fibroblast cell apoptosis induced by LPS through activation of the AKT/GSK3beta pathway. Biochem Moscow. 2017;82:606–612.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(–Delta Delta C(T)) method. Methods. 2001;25:402–408.
- Ding XM, Pan L, Wang Y, et al. Baicalin exerts protective effects against lipopolysaccharide-induced acute lung injury by regulating the crosstalk between the CX3CL1-CX3CR1 axis and NF-κB pathway in CX3CL1-knockout mice. Int J Mol Med. 2016;37:703.
- Grailer JJ, Canning BA, Kalbitz M, et al. Critical role for the NLRP3 inflammasome during acute lung injury. J Immunol. 2014;192:5974–5983.
- Khandelwal A, Bacolla A, Vasquez KM, et al. Long non-coding RNA: a new paradigm for lung cancer. Mol Carcinog. 2015;54:1235.
- Lu KH, Li W, Liu XH, et al. Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer. 2013;13:461.
- Terashima M, Tange S, Ishimura A, et al. MEG3 long noncoding RNA contributes to the epigenetic regulation of epithelial–mesenchymal transition in lung cancer cell lines. J Biol Chem. 2017;292:82.
- He JH, Han ZP, Liu JM, et al. Overexpression of long non-coding RNA MEG3 inhibits proliferation of hepatocellular carcinoma Huh7 cells via negative modulation of miRNA-664. J Cell Biochem. 2017;118:3713–3721.
- Golden RJ, Chen B, Li T, et al. An Argonaute phosphorylation cycle promotes microRNA-mediated silencing. Nature. 2017;542:197–202.
- Zhang D, Li Z, Zhang Y, et al. miR-4262 promotes the proliferation of human cutaneous malignant melanoma cells through KLF6-mediated EGFR inactivation and p21 upregulation. Oncol Rep. 2016;36:3657–3663.
- Bao H, Gao F, Xie G, et al. Angiotensin-converting enzyme 2 inhibits apoptosis of pulmonary endothelial cells during acute lung injury through suppressing MiR-4262. Cell Physiol Biochem. 2015;37:759–767.
- Ghaleb AM, Yang VW. Krüppel-like factor 4 (KLF4): what we currently know. Gene. 2017;611:27–37.
- Hu W, Jia Y, Yu Z, et al. KLF4 downregulates hTERT expression and telomerase activity to inhibit lung carcinoma growth. Oncotarget. 2016;7:52870.
- Zhao J, Chao C, Guo M, et al. MicroRNA-7 deficiency ameliorates the pathologies of acute lung injury through elevating KLF4. Front Immunol. 2016;7:389.
- Li C, Hu Y, Deng X, et al. Anti-apoptotic roles of MicroRNA-21 via activating PTEN/PI3K/Akt pathway in lipopolysaccharide-induced acute lung injury. Int J Clin Exp Pathol. 2017;10:4339–4347.
- Wang D, Li CY, Jin T, et al. Regulation of ENaC-mediated alveolar fluid clearance by insulin via PI3K/Akt pathway in LPS-induced acute lung injury. Respir Res. 2012;13:29.
- Zhao J, Yu H, Liu Y, et al. Protective effect of suppressing STAT3 activity in LPS-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2016;311:L868.
- Li C, Bo L, Li P, et al. Losartan, a selective antagonist of AT1 receptor, attenuates seawater inhalation induced lung injury via modulating JAK2/STATs and apoptosis in rat. Pulmon Pharmacol Ther. 2017;45:69–79.