
Abstract
The over-expanding role of lncRNA myocardial infarction associated transcript (MIAT) in various human diseases has been recently revealed. This study attempted to see the role of MIAT in a cell model of osteoarthritis (OA). ATDC5 cells were subjected to lipopolysaccharides (LPS) to mimic a cell model of OA. The effects of MIAT on the model were tested by performing CCK-8 assay, flow cytometry, qRT-PCR, western blot and ELISA. The downstream miRNA and signalling pathways were studied by utilizing qRT-PCR and western blot. Transfection of ATDC5 cells with the shRNA specific against MIAT significantly attenuated LPS-evoked apoptosis and cytokines release. At the meantime, the viability loss and the cleavage of caspases were ameliorated as well. MIAT overexpressed lead to the opposite result. Further, miR-132 was found to be negatively regulated by MIAT. The protective effects of MIAT silence were flattened when miR-132 expression was suppressed. Besides that the inhibitory effects of MIAT silence on LPS-evoked NF-κB and JNK activation were eliminated by miR-132 silence. This study illustrated that silence of MIAT protected ATDC5 cells against LPS challenge. The chondroprotective effects of MIAT silence may be via up-regulation of miR-132 and inhibition of NF-κB and JNK pathways.
Introduction
Osteoarthritis (OA) is a common disease occurred in the elder that seriously affects the quality of life among elderly people and costs huge medical expenses [Citation1]. With the ageing population gradually increased, the morbidity of OA is increased accordingly [Citation2]. Until now, no such drug has shown efficacy at slowing or reversing joint damage during OA progression in humans. Drug therapy mainly aims at relieving pain [Citation3], which necessitating joint replacement as a therapeutic option [Citation4]. Surgical therapy is a final choice and cannot solve articular cartilage degeneration. Thereby, improving our understanding of the mechanisms which drive the initiation and development of OA will be beneficial to prevent or treat this disease.
Long non-coding RNAs (lncRNAs) are a heterogeneous class of transcripts with a minimum length of 200 bases and without protein-coding potentials. A large number of studies have reported that lncRNAs are crucial players in human diseases via regulating genes which are essential in cellular biological processes. A variety of lncRNAs are studied, which highlighted the importance of lncRNAs in the pathogenesis of OA [Citation5,Citation6]. For the selected example, lncRNA TUG1 was able to promote chondrocyte extracellular matrix degradation during OA [Citation7]. Likewise, lncRNA PMS2L2 could protect chondrogenic cells against lipopolysaccharide (LPS) stimulation [Citation8]. LncRNA myocardial infarction associated transcript (MIAT), also known as RNCR2 or Gomafu, was first reported to be a functional RNA which confers risk of myocardial infarction [Citation9]. From then on, the over-expanding role of MIAT in various diseases has been revealed, like neurological disorders [Citation10], neovascular diseases [Citation11] and human cancers [Citation12]. Moreover, MIAT expression was closely associated with the number of lymphocytes, neutrophils and platelets, which linked MIAT with inflammatory response [Citation13]. However, the functional role of MIAT in inflammatory diseases is largely unknown.
microRNAs (miRNAs) are a large family of small (∼22 nucleotides) non-coding RNAs. Studies have evidenced miRNAs as important contributors in regulating osteogenic and chondrogenic differentiation and proliferation [Citation14,Citation15], which eventually influence the catabolism and anabolism of bone and cartilage [Citation16]. miR-132 is one of such miRNAs that has been identified as a regulator in chondrogenic differentiation [Citation17]. It is highly expressed in pristane-induced arthritis [Citation18,Citation19], suggesting miR-132 as a promising therapeutic target for treating OA. Besides that, a previous study has demonstrated miR-132 as a downstream effector of MIAT, via which MIAT exerted its oncogenic function [Citation20].
In this study, in vitro effects of MIAT silence on a cell model of OA which made by stimulating ATDC5 cells with LPS were studied to evaluate the role of MIAT in OA. In addition, the regulatory role of MIAT in miR-132 expression, as well as its downstream signalling, was explored to further reveal the underlying mechanisms.
Materials and methods
Cell
ATDC5 cells (European Collection of Cell Cultures, Salisbury, UK) were cultured in DMEM/HamF12 medium (Sigma-Aldrich, St. Louis, MO) supplemented with 5% FBS (Gibco, Grand Island, NY). The cells were routinely cultured in 5% humidified incubator at 37 °C and culture medium was renewed every other day. To make inflammatory injury, cells were treated by LPS (from Escherichia coli O111:B4, Sigma-Aldrich) for 12 h.
Transfection
pc-MIAT for expression of full-legend of MIAT was constructed by inserting MIAT sequences into pcDNA3.1 plasmid (Invitrogen, Carlsbad, CA). sh-MIAT, a shRNA specific for MIAT, was designed and synthesized by GenePharma (Shanghai, China). pcDNA3.1-empty vector and a non-targeting shRNA (sh-NC) served as blank controls for pc-MIAT and sh-MIAT. miR-132 inhibitor and the scrambled NC were from GenePharma. The vectors and oligonucleotides were transduced into ATDC5 cells with the mediation of lipofectamine 3000 (Invitrogen).
CCK-8 assay
Post-transfection, cells were placed into 96-well plates and cell viability was assessed by CCK-8 kit (Dojindo Molecular Technologies, Kyushu, Japan) following LPS stimulation. OD-values at 450 nm were recorded using a Bio-Rad ELISA reader (Hercules, CA).
Apoptosis assay
Post-transfection, cells were stimulated by LPS at 6-well plates, and then the apoptotic cells were stained by Annexin V-FITC Apoptosis Detection Kit (Beyotime, Shanghai, China). The rate of apoptotic cells was analyzed by a FACS can (Beckman Coulter, Fullerton, CA).
ELISA
The transfected cells were treated by LPS in 24-well plates, after which the culture supernatant was collected for use in ELISA. Concentrations of IL-6, IL-8, TNF-α and MCP-1 in culture supernatant were determined using the corresponding ELISA kits purchased from Abcam (Cambridge, MA).
qRT-PCR
Post-transfection and LPS stimulation, total RNAs were extracted by Trizol reagent (Invitrogen). To test the expression of IL-6, IL-8, TNF-α, MCP-1 and MIAT, PrimeScript™ RT Master Mix and TB Green Fast qPCR Mix both from Takara (Dalian, China) were utilized. To test the expression of miR-132, Mir-X™ miRNA First Strand Synthesis Kit and Mir-X™ miRNA qRT-PCR TB Green™ Kit both from Takara were utilized. GAPDH and U6 snRNA served as internal controls. Data was calculated according to 2−ΔΔCT method.
Western blot
Post-transfection and LPS stimulation, total proteins were isolated by RIPA lysis buffer (Beyotime, Shanghai, China). The primary antibodies used in this experimental system were as follows. Anti-caspase-3 (orb378617), anti-cleaved-caspase-3 (orb106556), anti-caspase-9 (orb135175), anti-cleaved-caspase-9 (orb227889), anti-PARP (orb526607), anti-cleaved-PARP (orb106557), anti-IL-6 (orb6210), anti-IL-8 (orb39299), anti-TNF-α (orb475251), anti-MCP-1 (orb97456), anti-p65 (orb344389), anti-p-p65 (orb14753), anti-IκBα (orb338946), anti-p-IκBα (orb99312), anti-JNK (orb38050), anti-p-JNK (orb184488), and anti-β-actin (orb86987) all from Biorbyt (San Francisco, CA). After incubation with secondary antibodies, the target bands were developed using ECL method.
Statistics
All results presented as mean ± SD. Statistical analysis was carried out in SPSS 19.0 software (Chicago, IL) using Student t-test or ANOVA combined with Duncan post-hoc test. A p value of <.05 was considered as statistical difference.
Results
ATDC5 cells injury evoked by LPS
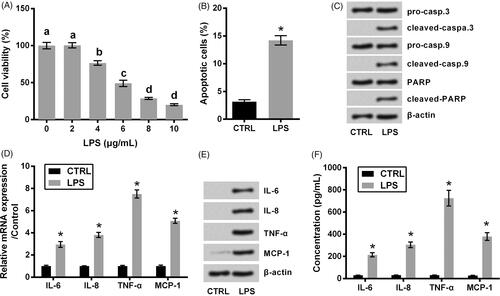
LPS with concentrations ranged from 2 to 10 μg/mL was utilized to injury ATDC5 cells, and the non-treated cells served as control. displayed that the viability of ATDC5 cells was gradually declined with increasing amount of LPS (p < .05). Considering 6 μg/mL LPS suppressed the viability down to about a half, 6 μg/mL was considered as an LPS-treating condition for use in the following experiments. displayed that LPS evoked a noteworthy apoptotic death in ATDC5 cells (p < .05). This result was coupled with the cleavage of caspase-3, caspase-9 and PARP (). Besides that, the expression and release of pro-inflammatory cytokines including IL-6, IL-8, TNF-α and MCP-1 were checked following LPS treatment. As seen in , these cytokines at mRNA and protein levels were dramatically increased by LPS (p < .05).
Figure 1. ATDC5 cells injury evoked by LPS. (A) The viability of ATDC5 cells was measured using CCK-8 assay, after treating with various concentrations of LPS (n = 3). Different letters above the columns indicate the significant difference (ANOVA combined with Duncan post-hoc test). The cells were treated by 6 μg/mL LPS, after which (B) apoptotic cell rate, (C) expression of proteins associated with apoptosis, (D) mRNA and (E) protein levels of pro-inflammatory cytokines, as well as (F) the release of the cytokines were measured using flow cytometry, western blot, qRT-PCR and ELISA (n = 3). *p < .05 (Student t-test).
Protective function of MIAT silence against LPS challenge in ATDC5 cells
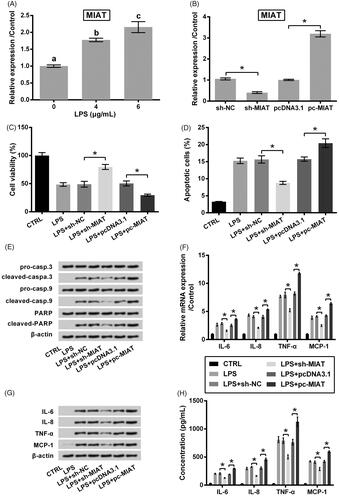
The expression of MIAT in LPS challenged ATDC5 cells was tested by qRT-PCR. revealed that MIAT expression was gradually increased with increasing amount of LPS (p < .05), which indicated that MIAT was sensitive to LPS response. Next, MIAT expression was suppressed or overexpressed by transfection with MIAT specific shRNA (sh-MIAT) or MIAT expressing vector (pc-MIAT) (both p < .05, ). Then, the role of MIAT in cell survival under LPS condition was tested. illustrated that transfection of cells with sh-MIAT attenuated LPS-evoked viability loss and apoptosis (p < .05). Of contrast, transfection of cells with pc-MIAT accelerated cell damage made by LPS (p < .05). Same trends were observed in the expression and release of pro-inflammatory cytokines. As seen in , the overproduction of cytokines made by LPs was alleviated by sh-MIAT while accelerated by pc-MIAT (p < .05).
Figure 2. Protective function of MIAT silence against LPS challenge in ATDC5 cells. (A) Relative expression of MIAT in ATDC5 cells was measured using qRT-PCR, after treating with various concentrations of LPS. Different letters above the columns indicate the significant difference. (B) Relative expression of MIAT was measured using qRT-PCR, after transfection with sh-MIAT, pc-MIAT or negative controls (sh-NC and pcDNA3.1). The transfected cells were then subjected to 6 μg/mL LPS, after which (C) cell viability, (D) apoptotic cell rate, (E) expression of proteins associated with apoptosis, (F) mRNA and (G) protein levels of pro-inflammatory cytokines, as well as (H) the release of the cytokines were measured using CCK-8 assay, flow cytometry, western blot, qRT-PCR and ELISA (n = 3). *p < .05 (ANOVA combined with Duncan post-hoc test).
Chondroprotective effects of MIAT silence through up-regulating miR-132
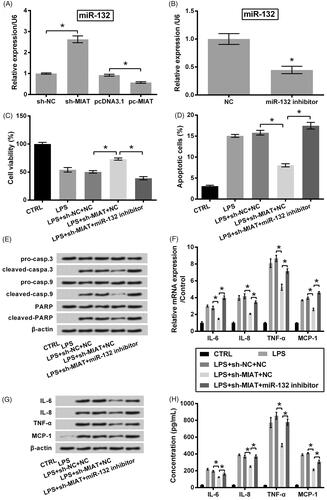
A previous study has demonstrated MIAT as a miR-132 sponge in colorectal cancer cells [Citation20]; thus, we, herein, studied whether MIAT silence exerted its chondroprotective function also via miR-132. qRT-PCR data in displayed a significant negative regulation between MIAT and miR-132 (p < .05). Next, miR-132 expression in ATDC5 cells was suppressed by inhibitor transfection (p < .05, ). We observed that the protective function of sh-MIAT silence was eliminated when miR-132 inhibitor was transfected into cell. To be specific, the effects of sh-MIAT on cell viability (p < 0.05, ), apoptosis (p < .05, ) and release of pro-inflammatory cytokines (p < .05, ) in LPS-injured ATDC5 cells were all flattened by miR-132 inhibitor.
Figure 3. Chondroprotective effects of MIAT silence through up-regulating miR-132. (A) Relative expression of miR-132 in ATDC5 cells was measured using qRT-PCR, after transfection with sh-MIAT, pc-MIAT or negative controls (sh-NC and pcDNA3.1). (B) Relative expression of miR-132 was measured using qRT-PCR, after transfection with miR-132 inhibitor or NC. The transfected cells were then subjected to 6 μg/mL LPS, after which (C) cell viability, (D) apoptotic cell rate, (E) expression of proteins associated with apoptosis, (F) mRNA and (G) protein levels of pro-inflammatory cytokines, as well as (H) the release of the cytokines were measured using CCK-8 assay, flow cytometry, western blot, qRT-PCR and ELISA (n = 3). *p < .05 (ANOVA combined with Duncan post-hoc test or Student t-test).
Suppressive effect of MIAT silence on NF-κB and JNK pathways via miR-132
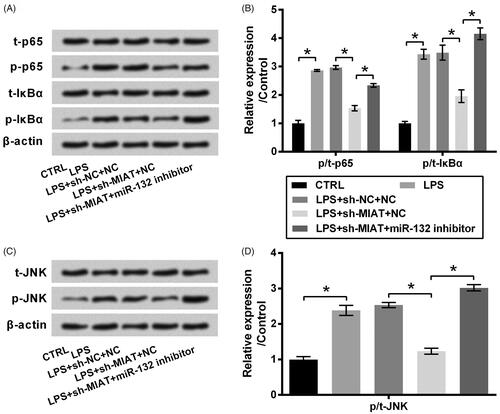
The signaling pathways of which MIAT silence exerted its chondroprotective function were also studied. Results in showed that LPS induced a significant activation of NF-κB and JNK pathways, as the phosphor/total (p/t) levels of p65, IκBα and JNK were noteworthy increased by LPS (p < .05). Transfection of cells with sh-MIAT attenuated LPS-evoked the activation of signalling pathways (p < .05); however, sh-MIAT was no longer in force when miR-132 inhibitor was transfected into ATDC5 cells (p < .05).
Figure 4. Suppressive effect of MIAT silence on the activation of NF-κB and JNK pathways via miR-132. ATDC5 cells were co-transfected with sh-MIAT and miR-132 inhibitor and then subjected to 6 μg/mL LPS. The phosphor/total (p/t) levels of (A,B) p65, IκBα and (C,D) JNK were measured using western blot (n = 3). *p < .05 (ANOVA combined with Duncan post-hoc test).
Discussion
Inflammation contributes to the symptoms and the progression of OA. Studies have suggested that disease-modifying interventions targeting inflammatory process may be effective for the prevention and treatment of OA [Citation21]. The present work attempted to reveal the role of MIAT in the inflammatory process of OA. To this end, MIAT expression was suppressed by shRNA-mediated silencing, and the effects on LPS-injured ATDC5 cells were analyzed. Our major findings are listed as follows. First, silencing MIAT effectively attenuated LPS-induced cell damage and the release of pro-inflammatory cytokines in ATDC5 cells. Second, miR-132 was a downstream effector of MIAT. MIAT functioned to LPS challenge in ATDC5 cells via regulating miR-132. Third, silencing MIAT suppressed inflammation-associated signalling (NF-κB and JNK) also via miR-132. These findings demonstrated MIAT as a contributor in LPS-induced inflammatory damage in ATDC5 cells.
MIAT was initially found to be pivotal in the pathogenesis of myocardial infarction [Citation9]. Beyond that, the importance of MIAT in other human diseases was widely studied. Its expression is aberrantly expressed in multiple diseases, like β-thalassemia [Citation22], breast cancer [Citation23] and schizophrenia [Citation24], indicating MIAT as an important regulator of these diseases. MIAT was also found to participate in the injury of various human body part, such as diabetic optic nerve [Citation25], myocardial tissue [Citation26] and renal tubules [Citation27]. Thereby, knockdown of MIAT may have clinical significance in preventing or treating human diseases [Citation13]. The present work demonstrated MIAT as a contributor in LPS-induced injury in ATDC5 cells. Despite the promoting roles of MIAT in cell apoptosis [Citation28] and inflammation [Citation13] in other experimental systems have been previously described, this work for the first time discovered these roles in a cell model of OA. The result provides in vitro evidence that silencing MIAT may have potential in treating OA.
The hypothesis of competing endogenous RNAs (ceRNAs) has been recently proposed to elucidate how lncRNAs regulate coding genes which are associated with cellular biological functions [Citation29]. To be specific, lncRNAs are able to sponge miRNAs, and thereby preventing the targeted mRNAs from degeneration by miRNAs. Thus, investigating the crosstalk between lncRNAs and miRNAs will be helpful for improving our understanding of lncRNAs. To date, several miRNAs have been reported to be sponged by MIAT, including miR-149-5p [Citation30], miR-150-5p [Citation31], miR-128-3p [Citation32], and miR-29c [Citation33]. Of note, miR-132, a key miRNA involved in chondrogenic differentiation [Citation17], was also found to be sponged by MIAT in colorectal cancer cells [Citation20]. In the current study, qRT-PCR data showed a negative regulation between MIAT and miR-132, which was consistence with previous finding indicated MIAT as a miR-132 sponge. Further experimental data suggested that silence of MIAT protected ATDC5 cells against LPS challenge via up-regulation of miR-132. Based on these evidence, we preliminary draw a conclusion that MIAT participated in OA pathogenesis at least partially through sponging miR-132.
NF-κB is a ubiquitous DNA-binding transcription factor. In general, NF-κB proteins are sequestered in the cytoplasm as inactive forms by their inhibitors like IκBα. However, upon the diverse stimulations such as TNF-α and LPS, NF-κB is activated and enters into the nucleus where it binds with their cognate DNA-binding sequences and regulates the expression of its downstream genes. The activated NF-κB and the stimulated pro-inflammatory cytokines lead to the onset and development of OA [Citation34]. Apart from NF-κB pathway, JNK pathway is also activated during OA and its activation contribute to cartilage degeneration [Citation35]. In response to stimulations like LPS, JNK pathway is activated and is able to induce cell apoptosis and inflammation [Citation36]. In the present study, the activation of these two pathways made by LPS was found to be attenuated by MIAT silence. Besides that, the inhibitory effects of MIAT silence on the pathways were flattened when miR-132 was down-regulated. These data suggested that MIAT silence suppressed LPS-evoked the activation of NF-κB and JNK pathways via regulating miR-132.
In conclusion, this study illustrated that silence of MIAT protected ATDC5 cells against LPS challenge. The chondroprotective effects of MIAT silence may be via up-regulation of miR-132 and deactivation of NF-κB and JNK pathways. However, further investigations are required to confirm this hypothesis in other experimental systems. The role of MIAT in other types of chondrocytes and animal model of OA will help to improve our findings. Also, the regulation of MIAT on miRNA network and the downstream signalling still needed to be further studied.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Related Research Data
References
- Beaudart C, Biver E, Bruyere O, et al. Quality of life assessment in musculo-skeletal health. Aging Clin Exp Res. 2018;30:413–418. May
- Sacitharan PK. Ageing and osteoarthritis. Subcell Biochem. 2019;91:123–159.
- Rodriguez-Merchan EC. Topical therapies for knee osteoarthritis. Postgrad Med. 2018;130:607–612.
- Santaguida PL, Hawker GA, Hudak PL, et al. Patient characteristics affecting the prognosis of total hip and knee joint arthroplasty: a systematic review. Can J Surg. 2008;51:428–436.
- Jeffries MA. Osteoarthritis year in review 2018: genetics and epigenetics. Osteoarthr Cartil. 2019;27:371–377.
- Jiang SD, Lu J, Deng ZH, et al. Long noncoding RNAs in osteoarthritis. Joint Bone Spine. 2017;84:553–556.
- Tang LP, Ding JB, Liu ZH, et al. LncRNA TUG1 promotes osteoarthritis-induced degradation of chondrocyte extracellular matrix via miR-195/MMP-13 axis. Euro Rev Med Pharmacol Sci. 2018;22:8574–8581.
- Li X, Yu M, Chen L, et al. LncRNA PMS2L2 protects ATDC5 chondrocytes against lipopolysaccharide-induced inflammatory injury by sponging miR-203. Life Sci. 2019;217:283–292.
- Ishii N, Ozaki K, Sato H, et al. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J Hum Genet. 2006;51:1087–1099.
- Barry G, Briggs JA, Vanichkina DP, et al. The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol Psychiatry. 2014;19:486–494.
- Yan B, Yao J, Liu JY, et al. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ Res. 2015;116:1143–1156.
- Crea F, Venalainen E, Ci X, et al. The role of epigenetics and long noncoding RNA MIAT in neuroendocrine prostate cancer. Epigenomics. 2016;8:721–731.
- Sun C, Huang L, Li Z, et al. Long non-coding RNA MIAT in development and disease: a new player in an old game. J Biomed Sci. 2018;25:23.
- Hodges WM, O'Brien F, Fulzele S, et al. Function of microRNAs in the osteogenic differentiation and therapeutic application of adipose-derived stem cells (ASCs). Int J Mol Sci. 2017;18:E2597.
- Yang Z, Li R, Ao J, et al. miR-1307-3p suppresses the chondrogenic differentiation of human adipose-derived stem cells by targeting BMPR2. Int J Mol Med. 2018;42:3115–3124.
- Cheng VK, Au PC, Tan KC, et al. MicroRNA and human bone health. JBMR Plus. 2019;3:2–13.
- Zhou X, Luo D, Sun H, et al. MiR-132-3p regulates ADAMTS-5 expression and promotes chondrogenic differentiation of rat mesenchymal stem cells. J Cell Biochem. 2018;119:2579–2587.
- Fernandes JG, Borrego A, Jensen JR. miRNA expression and interaction with genes involved in susceptibility to pristane-induced arthritis. J Immunol Res. 2018;2018:1928405.
- Murata K, Yoshitomi H, Tanida S, et al. Plasma and synovial fluid microRNAs as potential biomarkers of rheumatoid arthritis and osteoarthritis. Arthritis Res Ther. 2010;12:R86.
- Liu Z, Wang H, Cai H, et al. Long non-coding RNA MIAT promotes growth and metastasis of colorectal cancer cells through regulation of miR-132/Derlin-1 pathway. Cancer Cell Int. 2018;18:59.
- Liu-Bryan R, Terkeltaub R. Emerging regulators of the inflammatory process in osteoarthritis. Nat Rev Rheumatol. 2015;11:35–44.
- Fakhr-Eldeen A, Toraih EA. Long non-coding RNAs MALAT1, MIAT and ANRIL gene expression profiles in beta-thalassemia patients: a cross-sectional analysis. Hematology. 2019;24:308–317.
- Alipoor FJ, Asadi MH, Torkzadeh-Mahani M. MIAT lncRNA is overexpressed in breast cancer and its inhibition triggers senescence and G1 arrest in MCF7 cell line. J Cell Biochem. 2018;119:6470–6481.
- Li J, Zhu L, Guan F, et al. Relationship between schizophrenia and changes in the expression of the long non-coding RNAs Meg3, Miat, Neat1 and Neat2. J Psychiatr Res. 2018;106:22–30.
- Xu Y, Wang X, Zhang Y. Myocardial infarction-related transcripts (MIAT) participate in diabetic optic nerve injury by regulating heart shock protein 5 (HSPA5) via competitively binding to microRNA-379. Med Sci Monit. 2019;25:2096–2103.
- Liu Y, Wang T, Zhang M, et al. Down-regulation of myocardial infarction associated transcript 1 improves myocardial ischemia-reperfusion injury in aged diabetic rats by inhibition of activation of NF-kappaB signaling pathway. Chem Biol Interact. 2019;300:111–122.
- Zhou L, Xu DY, Sha WG, et al. Long non-coding MIAT mediates high glucose-induced renal tubular epithelial injury. Biochem Biophys Res Commun. 2015;468:726–732.
- Zhong X, Ma X, Zhang L, et al. MIAT promotes proliferation and hinders apoptosis by modulating miR-181b/STAT3 axis in ox-LDL-induced atherosclerosis cell models. Biomed Pharmacother. 2018;97:1078–1085.
- Yamamura S, Imai-Sumida M, Tanaka Y, et al. Interaction and cross-talk between non-coding RNAs. Cell Mol Life Sci. 2018;75:467–484.
- Ye ZM, Yang S, Xia YP, et al. LncRNA MIAT sponges miR-149-5p to inhibit efferocytosis in advanced atherosclerosis through CD47 upregulation. Cell Death Dis. 2019;10:138.
- Jin H, Jin X, Chai W, et al. Long non-coding RNA MIAT competitively binds miR-150-5p to regulate ZEB1 expression in osteosarcoma. Oncol Lett. 2019;17:1229–1236.
- Zhang C, Xie L, Liang H, et al. LncRNA MIAT facilitates osteosarcoma progression by regulating miR-128-3p/VEGFC Axis. IUBMB Life. 2019. doi: 10.1002/iub.2001
- Qu Y, Xiao H, Xiao W, et al. Upregulation of MIAT regulates LOXL2 expression by competitively binding MiR-29c in clear cell renal cell carcinoma. Cell Physiol Biochem. 2018;48:1075–1087.
- Rigoglou S, Papavassiliou AG. The NF-κB signalling pathway in osteoarthritis. Int J Biochem Cell Biol. 2013;45:2580–2584.
- Ge HX, Zou FM, Li Y, et al. JNK pathway in osteoarthritis: pathological and therapeutic aspects. J Recept Signal Transduct Res. 2017;37:431–436.
- Grynberg K, Ma FY, Nikolic-Paterson DJ. The JNK signaling pathway in renal fibrosis. Front Physiol. 2017;8:829.