
Abstract
Cellular senescence is strongly tied to vascular disease. The current study aims to examine ways that endothelial cellular senescence can be prevented and the mechanisms by which prevention of senescence occurs. Human umbilical vein endothelial cells were exposed to TNF-α to induce senescence; then salicin was administered in two doses – 50 and 100 µM – to establish a dose-dependent effect of salicin on SA-β-Gal, G1 cell cycle arrest, expression of p21 and PAI-1, p53 acetylation at K382, NRF2 and oxidative stress. NRF2 was examined as a mediating mechanism of salicin’s impact on cellular senescence and was found to account for salicin’s impact on SA-β-Gal, p21, PAI-1 and p53. Together, these results provide a compelling case that salicin has a substantial impact on numerous factors tied to cellular senescence in human endothelial cells. Thus, treatment with salicin may hold promise as a means of preventing aging-related vascular disease. Furthermore, salicin appears to operate via a functional pathway that is different from that affected by anti-inflammatory drugs (e.g. aspirin).
Introduction
Endothelial dysfunction plays a key role in the pathogenesis of atherosclerosis. In a young, healthy body, cells regenerate, not only healing from injury but also replenishing the cell population to cope with everyday wear-and-tear. Cellular senescence causes specific cells to stop replenishing [Citation1] and is also one of the major mechanisms of aging and health declines associated with aging. In particular, cellular senescence is strongly tied to age-related endothelial dysfunction and vascular disease which can emerge as a result [Citation2]. Since atherosclerotic cardiovascular diseases are the main cause of death worldwide [Citation3], and since endothelial cellular senescence is a key risk factor for vascular disease, it is worthwhile to examine methods by which endothelial cellular senescence can be prevented.
One key precursor to premature senescence is inflammation, and specifically, the inflammatory cytokine TNF-α [Citation4]. Exercise has been documented as being effective for reducing inflammation [Citation5], and previous research has found exercise to be an effective intervention to reduce endothelial senescence [Citation5]. However, exercise is only effective when practiced regularly, and adherence to exercise programmes is notoriously low among the elderly [Citation6], especially those living with chronic physical and psychological conditions [Citation7]. Medications known as TNF blockers [Citation8] are designed to inhibit TNF-α. In general, however, due to the safety and quality of life considerations, TNF inhibitors are indicated only for autoimmune diseases and other chronic medical conditions. TNF inhibitors are associated with increased risk of serious infections, such as tuberculosis and listeriosis, as well as increased risk of lymphoma, which is fatal [Citation9]. As a result, TNF inhibitors are an unlikely candidate for prophylactic treatment.
Salicin is a plant-based derivative that can be found in willow bark or in the stems and roots of Alangium chinense, an east and southeast Asian shrub [Citation10,Citation11]. It is a precursor to acetylsalicylic acid and has documented anti-inflammatory properties [Citation12], including an inhibitory effect on TNF-α [Citation13]. Whereas TNF-α inhibitors are typically administered via injection, salicin is ingested [Citation14] and has no noted serious side effects [Citation15,Citation16], fewer even than its synthetic counterpart, aspirin, and other non-steroidal anti-inflammatory drugs (NSAIDs), all of which can damage the gastrointestinal mucosa [Citation17].
In this study, we explore the potential applications of salicin for the amelioration of biological precursors to cardiovascular disease. We were particularly interested in the mechanisms by which salicin may prevent cellular senescence in endothelial cells, as little is known about this process. Specifically, we evaluated the effects of salicin on a number of variables when TNF-α was introduced to human epithelial cells. SA-β-Gal is a biomarker of senescence and has been identified as an indicator of aging [Citation18]. Salicin is hypothesized to inhibit SA-β-Gal activity. Conversely, the progression from the G1 phase of the cell cycle to the S phase is essential to cell proliferation [Citation19], and salicin is hypothesized to prevent disruption of the cell cycle. Expression of p21 [Citation20] and PAI-1 [Citation21] are tied to senescence in vascular smooth muscle cells (VSMCs) and chronic vascular disease. Furthermore, p53 acetylation at K382 induces senescence [Citation22] by promoting the expression of p21 [Citation23]. Salicin is expected to reduce the expression of all three. Nuclear factor erythroid-2 (NRF2) is a transcription factor that serves as a protective mechanism against free radical toxicity, oxidative insults, and threats to cell integrity [Citation24]. NRF2 is the mechanism by which exercise reduces vascular senescence, and similarly, salicin is expected to promote the nuclear translocation of NRF2. Lastly, we examined the capacity of salicin to decrease oxidative stress, which induces inflammation and leads to endothelial dysfunction. Lastly, we tested the hypothesis that the effects of salicin are mediated by NRF2. Together, these results provide a compelling case that salicin has a substantial impact on numerous factors tied to cellular senescence in human endothelial cells.
Materials and methods
Cell culture and treatment
HUVECs were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in an F12 K medium with a mixture of 10% FBS and 0.1% antibiotics. Cells were incubated with TNF-α (10 ng/mL) or salicin (50 and 100 μM) for 48 h.
SA-β-gal staining
HUVECs were plated into 96-well plates. After the necessary stimulation, cells were fixed and washed three times with ice-cold phosphate-buffered saline (PBS). Then, cells were incubated with the β-galactosidase staining reagent and maintained at 37 °C overnight. The staining was visualized.
Real-time PCR analysis
Total cellular RNA was extracted from HUVECs using Qiazol reagent (Qiagen, Germantown, MD). Purified RNA (1 μg) and a First Strand cDNA Synthesis Kit were used for reverse transcription reaction to synthesize cDNA. A commercial SYBR Green Master Mix (Bio-Rad, Hercules, CA), 1 μL synthesized cDNA and 0.4 μM primer pairs were used for real-time PCR. The data were quantified using the 2−ΔΔCT method with GAPDH as a reference.
Western blot analysis and immunoprecipitation
HUVECs from the control and treatment groups were lysed in RIPA buffer. After centrifugation, the protein concentration of the supernatant was assayed using a commercial BCA kit (Sigma-Aldrich, St. Louis, MO). Then, proteins (20 μg) were subjected to 10% SDS-PAGE and transferred to PVDF membranes. After blocking, sequential incubation with primary antibodies and HRP-linked secondary antibody, blots were detected with enhanced chemiluminescence. Immunoprecipitation assay was used to determine K382 acetylation of p53. Briefly, equal amounts of lysate of HUVECs (500 μg) from each group were used to incubate with p53 polyclonal antibody (5 µg) overnight at 4 °C. Then, 10% protein A (50 µL) was added and maintained for 30 min at 4 °C. Immunoprecipitants were assayed using western blot [Citation25].
Flow cytometry
The cell cycle of HUVECs under different conditions was assessed using flow cytometry. After the indicated stimulation, cells were washed and fixed with 70% ethanol. After being permeabilized with 0.1% Triton X-100 in Tween-20, cells were probed with propidium iodide (PI). Cell cycle was analyzed through flow cytometry.
Measurement of reactive oxygen species (ROS)
Oxidative stress in HUVECs was assessed by measuring the levels of ROS with 2,7-Dichlorodihydrofluorescein diacetate stain (DCFH-DA). DCFH-DA was diluted to 10 µM in serum-free F12 K medium. After indicated stimulation, diluted DCFH-DA was added to the cell culture for 30 min at 37 °C. After three washes, fluorescent images were detected using a fluorescence microscope.
Cell viability measurement
After the indicated treatment, cell viability was examined using an MTT assay. Cells were loaded with 0.2 mg/ml MTT and cultured for 60 min at 37 °C. The reaction products were solubilized in DMSO. OD value was determined at 560 nm to index cell viability.
Statistical analysis
Data are displayed as means ± SEM. Statistical analysis was performed using one-way ANOVA. A p-value ≤ .05 was considered statistically significant.
Results
Salicin inhibited TNF-α-induced increase in SA-β-Gal activity
First, we investigated the cytotoxicity of salicin in HUVECs. The results of MTT assay in indicate that when the concentration of salicin was less than 100 μM, it did not exert a significant impact on the cell viability of HUVECs. However, 500 μM salicin induced slight cytotoxicity of HUVECs. Therefore, concentrations of 50 and 100 μM salicin were used in this study. Because TNF-α is known to increase SA-β-Gal activity, and SA-β-Gal is a biomarker of cellular senescence, we hypothesized that treatment with salicin would prevent the increase in SA-β-Gal activity typically caused by TNF-α. As seen in , when endothelial cells were exposed to 10 ng/mL TNF-α, those not treated by salicin displayed significantly greater cellular senescence after 48 h, as compared to cells that were not exposed to TNF-α. In contrast, salicin prevented senescence in a dose-dependent manner; endothelial cells treated with 50 and 100 µM of salicin showed proportionately less cellular senescence, with the 100 µM assay relative SA-β-Gal level of the 100 µM assay approaching that of the assay not treated with TNF-α.
Figure 1. Salicin inhibited TNF-α-caused elevation of SA-β-Gal activity in HUVECs. Cells were incubated with TNF-α (10 ng/mL) or salicin (50 and 100 μM) for 48 h. (A) Cellular senescence in HUVECs was measured by SA-β-Gal staining. (B) Percentages of SA-β-gal positive cells (*,#,$p < .01 vs. previous group).
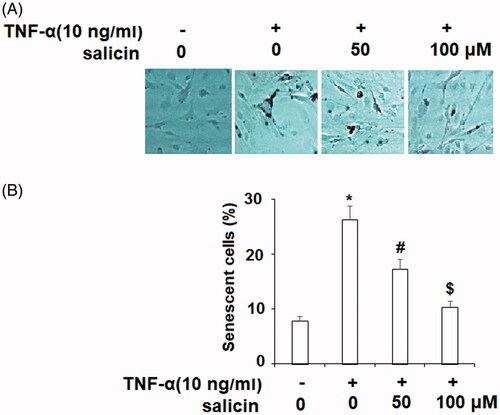
Salicin prevented TNF-α-induced cell cycle arrest in the G1 phase
TNF-α disrupts the cell cycle in the G1 phase, thereby halting cell proliferation in endothelial cells [Citation26]. Salicin was hypothesized to prevent TNF-α-induced cell cycle disruption at G0/G1, allowing normal cell proliferation to continue in endothelial cells. As seen in , cell cycle analysis indicated that whereas TNF-α-affected cells showed a higher level of cell cycle dysfunction compared to cells that were not exposed to TNF-α, salicin demonstrated a protective effect against cell cycle disruption at G0/G1. There was a dose-response relationship such that endothelial cells treated with 50 µM showed less cell cycle disruption than the untreated assay, and cells treated with 100 µM of salicin showed levels of cycle disruption similar to the assay not treated with TNF-α.
Figure 2. Salicin prevented TNF-α-caused cell cycle arrest in the G1 phase in HUVECs. Cells were cultured with TNF-α (10 ng/mL) or salicin (50 and 100 μM) for 48 h. Cell cycle analysis was measured using FACS (*,#,$p<.01 vs. previous group).
While TNF-α halts the cell cycle in G0/G1, it is known to reduce barriers to cell reproduction in both S and G2/M phases, where unhealthy cells are normally inhibited from reproducing [Citation27]. As a result, progression through S and G2/M should be more fluid when TNF-α is present. Smaller, but statistically significant effects of TNF-α were seen on progress through S and G2/M phases in the expected direction, with salicin reducing the impact of TNF-α at 50 µM and eliminating it entirely at 100 µM.
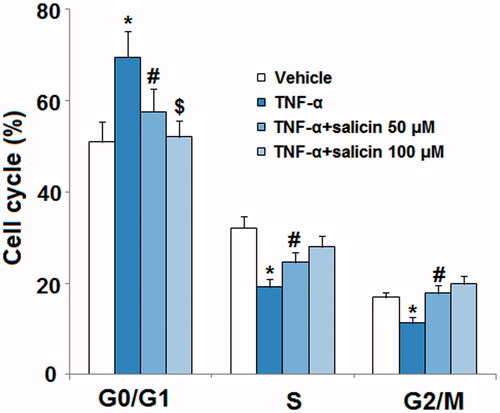
Salicin reduced TNF-α-induced expression of p21 and PAI-1
TNF-α promotes expression of p21 and PAI-1, both of which are tied to senescence in endothelial cells. As shown in , mRNA-level and protein-level analyses revealed that while TNF-α remarkably increased expression of p21 and PAI-1 in untreated endothelial cells, salicin somewhat reduced p21 and PAI-1 expression at the 50 µM dose, and significantly reduced p21 and PAI-1 expression at the 100 µM dose, showing a dose-response relationship.
Figure 3. Salicin reduced TNF-α-caused expression of p21 and PAI-1 in HUVECs. Cells were incubated with TNF-α (10 ng/mL) or salicin (50 and 100 μM) for 48 h. (A) mRNA levels of p21 and PAI-1. (B) Protein levels of p21 and PAI-1 (*,#,$p < .01 vs. previous group).
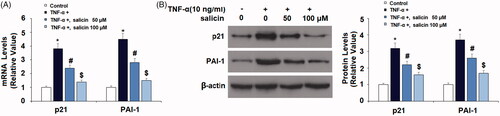
Salicin prevented TNF-α-induced K382 acetylation of p53 (ac-K382)
TNF-α significantly increased the level of total p53 which was reduced by salicin. Importantly, TNF-α induces K382 acetylation of p53, which, in turn, promotes the expression of p21 and PAI. As illustrated in , while TNF-α significantly increased the acetylation of p53 in untreated cells, salicin led to levels of p53 acetylation 34.2% lower than untreated cells at 50 µM and 58.5% lower than untreated cells at 100 µM, indicating a dose-response relationship.
Figure 4. Salicin prevented TNF-α-caused K382 acetylation of p53 (ac-K382). HUVECs were incubated with TNF-α (10 ng/mL) or salicin (50 and 100 μM) for 48 h. Acetylation of p53 was measured with immunoprecipitation (*,#,$p<.01 vs. previous group).
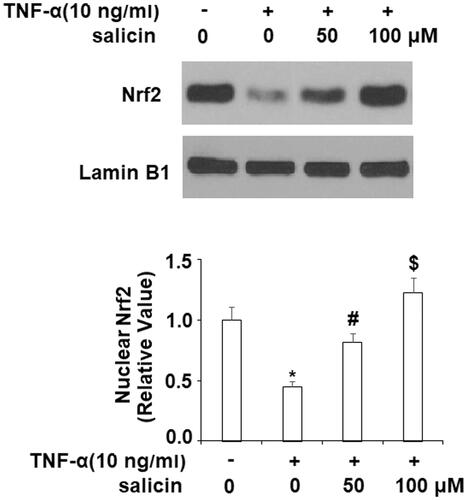
Salicin promoted nuclear translocation of NRF2
NRF2 is a transcription factor that protects against cellular senescence through multiple mechanisms. As illustrated in , while nuclear levels of NRF2 were reduced in the group exposed to TNF-α, replicating previous research that TNF-α hinders nuclear translocation of NRF2. Importantly, we found that salicin promoted nuclear translocation of NRF2.
Figure 5. Salicin promoted the nuclear translocation of NRF2. Cells were treated with TNF-α (10 ng/mL) or salicin (50 and 100 μM) for 48 h. Nuclear levels of NRF2 in HUVECs were measured (*,#,$p < .01 vs. previous group).
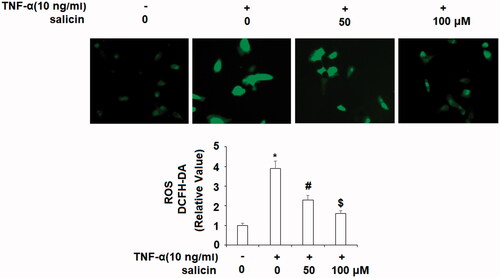
Salicin prevented TNF-α-induced oxidative stress
NRF2 has been identified as an anti-oxidative stress transcriptional factor in different cells and tissues. Oxidative stress induces inflammation and leads to vascular senescence; it is a key mechanism by which TNF-α leads to vascular disease. As displayed in , TNF-α induced a significant increase in oxidative stress, while salicin treatment moderately prevented oxidative stress at 50 μM, and more potently with a dose of 100 μM salicin showing a dose–response relationship.
Figure 6. Salicin prevented TNF-α- induced oxidative stress in HUVECs. Cells were incubated with TNF-α (10 ng/mL) or salicin (50 and 100 μM) for 48 h. Production of ROS was measured using DCFH-DA staining (*,#,$p<.01 vs. previous group).
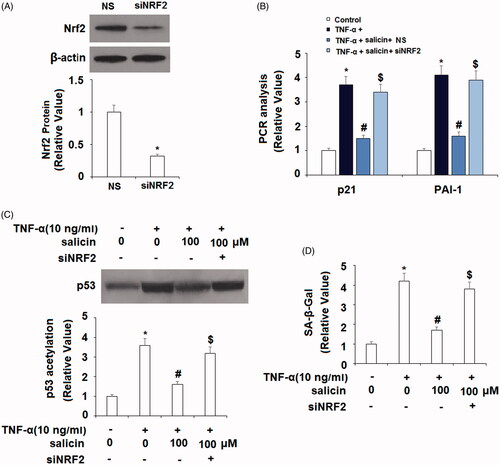
The effects of salicin are mediated by NRF2
We then conducted a mediation analysis (see ) to explore the role of NRF2 as the driving force behind the impact of salicin on cellular senescence through knockdown of NRF2 through small RNA transfection. Successful knockdown of NRF2 is shown in . Interestingly, inhibition of NRF2 abolished the inhibitory effects of salicin on the expression of p21 and PAI-1 (), p53 acetylation () and SA-β-Gal activity (). These findings suggest that salicin’s impact on NRF2 accounted for its impact on p21, PAI-1, p53 acetylation, and endothelial senescence.
Figure 7. Silencing of NRF2 abolished the effects of salicin in inhibiting cellular senescence in HUVECs. Cells were transfected with NRF2 siRNA. Twenty four hours later, cells were incubated with TNF-α (10 ng/mL) or 100 μM salicin for 48 h. NS: non-specific small RNA; siNRF2, NRF2 siRNA. (A) Successful knockdown of NRF2. (B) Silencing of NRF2 abolished the inhibitory effects of salicin on p21 and PAI-1 expression. (C) Immunoprecipitation assay revealed that knockdown of NRF2 abolished the inhibitory effects of salicin on p53 acetylation. (D) SA-β-Gal staining results demonstrate that silencing of NRF2 abolished the inhibitory effects of salicin in cellular senescence (*,#,$p<.01 vs. previous group).
Salicin inhibited H2O2-induced increase in cellular senescence
Cellular senescence is induced by ROS. To further confirm the beneficial capacity of salicin against cellular senescence, HUVECs were stimulated with H2O2 (100 μM) with or without salicin (50 and 100 μM) for 24 h. The results of SA-β-gal staining in indicate that stimulation with H2O2 significantly increased the percentage of SA-β-gal-positive cells, which was greatly reduced by salicin. Consistently, salicin inhibited H2O2-induced expression of p21 and PAI-1 (). These findings suggest that salicin possesses a protective effect against H2O2-induced cellular senescence in HUVECs.
Discussion
In this article, we examined salicin, which has anti-inflammatory properties but is not an NSAID, as a means of preventing the cellular senescence that can result from inflammation. Importantly, NSAIDS, steroids and other treatments for inflammation have side effects from long-term use, so continuing research is needed to explore alternative therapeutic agents that may facilitate the reduction of inflammation. We discovered that salicin treatment, when administered to endothelial cells being impacted by the proinflammatory factor TNF-α, has a significant inhibitory impact on cellular senescence, translocation of NRF2 and oxidative stress. Perhaps most interestingly, we examined the underlying mechanism of these relationships and found that NRF2 accounted for much of salicin’s effects on cellular senescence in endothelial cells. NRF2, therefore, can be considered a key mechanism underlying the effectiveness of salicin as a preventive treatment for vascular disease.Brief human trials have found salicin to have minimal side effects comparable to, or perhaps even less than, aspirin and other NSAIDs, specifically when it comes to gastrointestinal side-effects [Citation15,Citation16]. However, as many of the negative effects of NSAID and steroid treatment are long-term, it is important for future research to explore the long-term impact of taking salicin, both in terms of its preventive effects and its potential side effects. Nevertheless, the current results provide a rationale for doing further testing of the potential efficacy of salicin in this domain.
While previous studies have documented the benefit of salicin in reducing inflammation in humans with inflammatory diseases such as osteoarthritis, no studies have examined salicin’s potential role in preventing vascular disease. While salicin is a precursor to aspirin, and aspirin (along with other NSAIDs) is a widely-used tool for preventing vascular disease, salicin differs from aspirin in several key ways and is thought to operate by a different, broader mechanism of action than aspirin [Citation17]. This study suggests that despite the differences between salicin and aspirin, salicin has potential as a prophylactic approach against vascular disease. It is important to note that while the primary mechanisms underlying the impact of aspirin on cardiovascular disease revolve around the suppression of platelet aggregation in COX platelets and suppression of prostaglandin production [Citation28], the current study shows that salicin operates by a different mechanistic chain, specifically, through nuclear translocation of NRF2. The potential benefits of salicin for the prevention of vascular disease, therefore, are unique to salicin, rather than merely a result of salicin’s molecular relation to aspirin.
Supplemental Material
Download ()Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Lynch MD. How does cellular senescence prevent cancer? DNA Cell Biol. 2006;25:69–78.
- Hadi HAR, Carr CS, Al Suwaidi J. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vasc Health Risk Manag. 2005;1:183–198.
- Barquera S, Pedroza-Tobías A, Medina C, et al. Global overview of the epidemiology of atherosclerotic cardiovascular disease. Arch Med Res. 2015;46:328–338.
- Li P, Gan Y, Xu Y, et al. The inflammatory cytokine TNF-α promotes the premature senescence of rat nucleus pulposus cells via the PI3K/Akt signaling pathway. Sci Rep. 2017;7:42938.
- Dimitrov S, Hulteng E, Hong S. Inflammation and exercise: inhibition of monocytic intracellular TNF production by acute exercise via β 2 -adrenergic activation. Brain Behav Immun. 2017;61:60–68.
- Chao D, Foy CG, Farmer D. Exercise adherence among older adults. Control Clin Trials. 2000;21:212S–217S.
- Garmendia ML, Dangour AD, Albala C, et al. Adherence to a physical activity intervention among older adults in a post-transitional middle-income country: a quantitative and qualitative analysis. J Nutr Health Aging. 2012;17:466–471.
- Lis K, Kuzawińska O, Bałkowiec-Iskra E. State of the art paper tumor necrosis factor inhibitors-state of knowledge. Arch Med Sci. 2014;6:1175–1185.
- Jain A, Singh JA. Harms of TNF inhibitors in rheumatic diseases: a focused review of the literature. Immunotherapy. 2013;5:265–299.
- Zhang Y, Liu YB, Li Y, et al. Sesquiterpenes and alkaloids from the roots of Alangium chinense. J Nat Prod. 2013;76:1058–1063.
- Aboelsoud N. Herbal medicine in ancient Egypt. J Med Plants Res. 2010;4:82–86.
- Khayyal M, El-Ghazaly M, Abdallah D, et al. Mechanisms involved in the anti-inflammatory effect of a standardized willow bark extract. Arzneimittelforschung. 2011;55:677–687.
- Bonaterra G, Heinrich E, Kelber O, et al. Anti-inflammatory effects of the willow bark extract STW 33-I (Proaktiv®) in LPS-activated human monocytes and differentiated macrophages. Phytomedicine. 2010;17:1106–1113.
- Smithson J, Kellick K, Mergenhagen K. Nutritional modulators of pain in the aging population. Amsterdam (Netherlands): Elsevier Science; 2017. p. 191–198.
- Schmid B, Lüdtke R, Selbmann HK, et al. Efficacy and tolerability of a standardized willow bark extract in patients with osteoarthritis: randomized placebo-controlled, double-blind clinical trial. Phytother Res. 2001;15:344–350.
- Shara M, Stohs SJ. Efficacy and safety of white willow bark (Salix alba) extracts. Phytother Res. 2015;29:1112–1116.
- Vlachojannis J, Magora F, Chrubasik S. Willow species and aspirin: different mechanism of actions. Phytother Res. 2011;25:1102–1104.
- Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367.
- Bertoli C, Skotheim JM, Bruin R. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol. 2013;14:518–528.
- Kim HS, Cho HJ, Cho HJ, et al. The essential role of p21 in radiation-induced cell cycle arrest of vascular smooth muscle cell. J Mol Cell Cardiol. 2004;37:871–880.
- Luo M, Ji Y, Luo Y, et al. Plasminogen activator inhibitor-1 regulates the vascular expression of vitronectin. J Thromb Haemost. 2017;15:2451–2460.
- Reed S, Quelle D. p53 Acetylation: regulation and consequences. Cancers (Basel). 2014;7:30–69.
- Zhao Y, Lu S, Wu L, et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21Waf1/Cip1. Mol Cell Biol. 2006;26:2782–2790.
- Satta S, Mahmoud AM, Wilkinson FL, et al. The role of Nrf2 in cardiovascular function and disease. Oxid Med Cell Longev. 2017;2017:1–18.
- Song W, Zhang Y, Wang J, et al. Antagonism of cysteinyl leukotriene receptor 1 (cysLTR1) by montelukast suppresses cell senescence of chondrocytes. Cytokine. 2018;103:83–89.
- Spyridopoulos I, Principe N, Krasinski KL, et al. Restoration of E2F expression rescues vascular endothelial cells from tumor necrosis factor-alpha-induced apoptosis. Circulation. 1998;98:2883–2890.
- Faurschou A. Role of tumor necrosis factor-α in the regulation of keratinocyte cell cycle and DNA repair after ultraviolet-B radiation. Dan Med Bull. 2010;57:B4179.
- Ittaman SV, VanWormer JJ, Rezkalla SH. The role of aspirin in the prevention of cardiovascular disease. Clin Med Res. 2014;12:147–154.