
Abstract
Emodin (EMO) possesses extensive pharmacological activities, which has been proven to exert the protective impact in diverse nervous system diseases. Nonetheless, whether EMO emerges a neuro-protective activity in hypoxic-evoked ischemic brain injury is still further probed. The intention of the research is to disclose whether EMO emerges neuro-protective activity in hypoxic-evoked ischemic brain injury. PC-12 received hypoxia administration, and then cell viability, apoptosis and autophagy were estimated. After EMO disposition, the above-involved cellular processes were evaluated again. MiR-25 functions in EMO-affected cells were also estimated. The interrelation between miR-25 and neurofilament light-chain polypeptide gene (NEFL) and the conceivable roles of NEFL in hypoxia-disposed cells were investigated. The latent mechanism was uncovered by mTOR and Notch pathways determination. Hypoxia triumphantly triggered apoptosis and autophagy, but EMO repressed these functions in PC-12 cells. Increased miR-25 was induced by EMO, and inhibited miR-25 abated the impacts of EMO on hypoxia-disposed PC-12 cells. NEFL as a neoteric target gene of miR-25 was predicated, and overexpressed NEFL annulled the functions of EMO in hypoxia-injured cells. EMO activated mTOR and Notch pathways through repressing NEFL. The investigations corroborated that EMO weakened hypoxia-triggered injury via elevating miR-25 by targeting NEFL in PC-12 cells.
Introduction
Cerebral ischemia is also called as brain ischemia or cerebrovascular ischemia, which refers to a situation that the blood supply to the brain is insufficient to satisfy the requirement of metabolism [Citation1,Citation2]. Ischemia can give rise to miscellaneous alterations in brain metabolism and energy crisis [Citation3]. Moreover, cerebral ischemia is usually discovered in the pathological processes of diversified neurological diseases, comprising cerebrovascular disease and brain tumours [Citation4]. The aetiology of cerebral ischemia is complex, which might be linked to cerebral arteriosclerosis [Citation5]. The impairments triggered by cerebral ischemia encompass vision, speaking and physical activity [Citation6,Citation7]. Currently, there is no valid management for cerebral ischemia, which can only be controlled by relevant drugs [Citation8]. It is generally known that cerebral ischemia can evoke hypoxic injury in brain nerve cells [Citation9]. Hence, to probe the molecular mechanism and mediation of hypoxic-triggered nerve cell injury is extremely important for remedying nerve injury after cerebral ischemia.
Emodin (EMO, 1,3,8-trihydroxy-6-methylanthraquinone) is a dominating anthraquinone derivative extracting from Rheum palmatum L. [Citation10]. EMO possesses comprehensive application in clinical practice, and the anti-tumour [Citation11], anti-inflammatory [Citation12], anti-bacterial [Citation13] and anti-fibrotic [Citation14] activities have been testified in all kinds of ailments. Increasing evidence disclosed that EMO exhibited the protective impacts in diverse nervous system diseases, encompassing neuropathic pain [Citation15] and Alzheimer’s disease [Citation16]. These explorations hinted the neuro-protective impacts of EMO on these diseases. Moreover, a compelling research uncovered that EMO gave play to a neuro-protective activity in hypoxic-evoked ischemic brain injury [Citation17]. Therefore, we further probed whether EMO could protect PC-12 cells against hypoxia-triggered injury.
MicroRNAs (miRNAs) are a sort of weeny RNAs absenting the ability to encode proteins, which join in controlling multiple biological processes [Citation18]. Actually, the researches focused on miRNAs are developing rapidly and increasing evidences have disclosed the importance of miRNAs in ischemic disease [Citation19]. Recent research has corroborated that EMO exerted the functions through regulating miRNAs. Evidence from Xiang et al. revealed that EMO could flatten sodium taurocholate-triggered pancreatic acinar cell injury via miR-30a-5p-mediated inflammatory signaling [Citation20]. Moreover, Pan et al. disclosed that EMO restrained transforming growth factor beta-2 (TGF-β2) via activating the forkhead box D3 (FOXD3)/miR-199a axis in ovarian cancer cells. These above-mentioned evidences hinted a potential relevancy between EMO and miRNAs. MiR-25 is a proverbially studied miRNA, which is certified to protect cardiomyocytes against oxidative damage through the mitochondrial calcium uniporter [Citation21]. Additionally, miR-25 exhibited the protective action to withstand hypoxia/reoxygenation (H/R)-triggered fibrosis and apoptosis in H9c2 cells [Citation22]. Nonetheless, whether miR-25 participates in governing the functions of EMO in hypoxia-damaged PC-12 cells remains dimness. Herein, the research probed the protective function of EMO in hypoxia-damaged PC-12 cells. In the meaning time, the involvements of miR-25 in this progress have been also investigated. The explorations might uncover the latent functions of EMO in cerebral ischemia.
Materials and methods
Cell culture and disposition
The experimental PC-12 cells attained from American Type Culture Collection (ATCC® CRL-1721™ Rockville, MD, USA) were fostered in ATCC-formulated RPMI-1640 medium (ATCC® 30–2001™) incorporating 10% heat-inactivated horse serum (ATCC® 30–2040™, Rockville, MD, USA) and 5% foetal bovine serum (FBS) (ATCC® 30–2020™, Rockville, MD, MSA). PC-12 cells were kept in an incubator with 5% CO2 and 95% air at 37 °C. PC-12 cells received hypoxia disposition, which were lasted for 12 h in a hypoxic incubator (94% N2, 5% CO2 and 1% O2, SANYO, Osaka, Japan). EMO was bought from Sigma (St Louis, MO, USA), meanwhile dissolved in DMSO (ATCC® 4-X™, Rockville, MD, USA) to configure diverse dosage solutions (0, 5, 10 and 20 μM). EMO at above-allocated concentration was utilized to dispose PC-12 cells for 12 h.
Cell transfection
The disparate expression vectors of miR-25 mimic and inhibitor and the neurofilament light-chain polypeptide gene (NEFL) overexpressed vector (pc-NEFL) were all synthesized by GenePharma (Shanghai, China). The NC mimic, NC inhibitor and pcDNA3.1 vectors served as the correlative blank controls. The above vectors were transfected into PC-12 cells via executing Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA) in accordance with its specification.
Assessment of cell viability
PC-12 cell viability was appraised via utilizing Cell Counting Kit-8 (CCK-8, Dojindo, Gaithersburg, MD, USA) after hypoxia and EMO administration with miR-25 inhibitor transfection. These PC-12 cells were fostered in 96-well plate for 12 h subsequently 10 μL CCK-8 solution was mixed into the culture plate and co-fostered for extra 2 h at 37 °C. The absorbance of the disposed and transfected cells was evaluated at 450 nm via adopting a Microplate Reader (Bio-Rad, Hercules, CA, USA).
Determination of cell apoptosis
After hypoxia and EMO administration with miR-25 inhibitor or pc-NEFL transfection, cell apoptosis were assessed through adopting annexin V-FITC/propidium iodide (PI) kit (Biosea, Beijing, China). The disposed and transfected PC-12 cells were rinsed twice with 4 °C phosphate buffered saline (PBS) and re-suspended in 400 μL 1 × binding buffer. Afterward, PC-12 cells were co-cultivated with 5 μL annexin V-FITC for 15 min absence of light, concurrently 5 μL PI was replenished for reaction for additional 15 min. Cell apoptosis was ultimately estimating by adopting flow cytometry instrument (FACSAria II, BD Biosiences, San Jose, CA, USA).
Dual luciferase reporter assay
PCR was executed to amplify the fragment from NEFL 3′UTR incorporating the predicted miR-25 binding site. The amplification products were then cloned into pmirGLO vector (Promega, Madison, WI, USA) to construct the reporter vector NEFL-wild-type (NEFL-wt). QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) was exploited to build NEFL-mutated-type (NEFL-mut). Afterward, the above-constructed vectors and miR-25 mimic or NC mimic were co-transfected into PC-12 cells, meanwhile Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA) was executed for luciferase activity assessment.
Real-time quantitative PCR (RT-qPCR)
We utilized TRIzol reagent (Invitrogen, Carlsbad, CA) for extraction of total RNA from disposed and transfected PC-12 cells. In line with the specification, PrimeScript 1st Strand cDNA Synthesis kit (TaKaRa, Siga, Japan) was employed for cDNA synthesis. According to Taqman microRNA RT Kit and Applied Biosystems™ TaqMan (ABI, Foster City, CA, USA) specification, real-time quantitative polymerase chain reaction (RT-qPCR) system was configured for PCR amplification. The concrete thermocycler conditions for RT-qPCR were initial denaturation at 95 °C for 15 s along with 30 cycles for 30 s at 95 °C, annealing at 61 °C for 5 s, extension at 72 °C for 15 s and the final extension at 72 °C for 10 min. U6 and β-actin were considered to be the internal references for testing miR-25 and NEFL expression. The correlative data were calculated by adopting the classical 2−ΔΔCt method [Citation23].
Western blot assay
After disposition and transfection, the protein specimens were prepared by radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime, Shanghai, China) incorporating protease inhibitor (Roche, San Francisco, CA, USA). BCA™ Protein Assay Kit (Beyotime, Shanghai, China) was adopted for total protein concentration assessment. Afterward, these proteins were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), in the meantime electro-transferred to nitrocellulose membranes (Millipore, Billerica, MA, USA). After sealing with 5% BSA, the interrelated primary antibodies were co-cultivated with the nitrocellulose membranes overnight at 37 °C. The primary antibodies incorporated Bax (ab53154), pro-Caspase-3 (ab32499), cleaved-Caspase-3 (ab49822), PARP (ab191217), cleaved-PARP (ab32064), Beclin-1 (ab207612), p62 (ab91526), LC3-II (ab192890), NEFL (ab223343), t-p70S6K (ab32529), p-p70S6K (ab59208), t-mTOR (ab32028), p-mTOR (ab137133), Notch-1 (ab52627), Notch-2 (ab137665) and β-actin (ab227387, Abcam, Cambridge, UK). Thereafter, the second primary (ab205718, Abcam, Cambridge, UK) was co-incubated with the nitrocellulose membranes for additional 1 h at circumambient temperature. The enhanced chemiluminescence (ECL) reagent (GE Healthcare, Braunschweig, Germany) was adopted for visualising the bands and the optical density was analysed by ImageJ software (Bio-Rad).
Statistical analysis
The results in the actual research were emerged as the mean ± standard deviation (SD). SPSS version 19.0 statistical software (IBM, Armonk, NY) was adopted for statistical analyses. Student t-test and analysis of variance (ANOVA) along with Tukey post-hoc test were exploited to analyse the data in two groups or multiple groups. A p < .05 was deemed as a result which possessed significant difference.
Results
Hypoxia triggered PC-12 cells apoptosis and autophagy
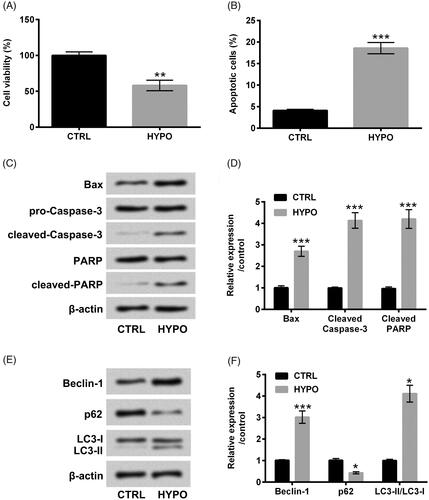
PC-12 cells received hypoxia administration for 12 h, and then the variations of PC-12 cells were evaluated. Contrasted with control group (cells under normaxia condition), cell viability was evidently reduced and cell apoptosis was simultaneously augmented under hypoxia disposition (p < .01 or p < .001, ). Additionally, western blot analytic consequences revealed that Bax, cleaved-Caspase-3 and cleaved-PARP protein levels were both elevated under hypoxia administration (p < .001, ). Outside of this, the enhancements of Beclin-1 (p < .001) and LC3-II (p < .05) as well as the inhibition of p62 (p < .05) were observed after hypoxia disposition (). The above discoveries suggested that hypoxia disposition successfully trigger cell apoptosis and autophagy in PC-12 cells.
Figure 1. Hypoxia triggered apoptosis and autophagy in PC-12 cells. After administration with hypoxia for 12 h, CCK-8 and flow cytometry were executed for (A) cell viability and (B) cell apoptosis determination. Western blot assay was carried out for (C and D) Bax, pro/cleaved-Cleaved-3, PARP and cleaved-PARP and (E and F) Beclin-1, p62 and LC3-I/LC3-II protein levels assessment. The three independent experiments were conducted. *p < .05, **p < .01 and * **p < .001.
EMO relieved hypoxia-triggered PC-12 cells apoptosis and autophagy
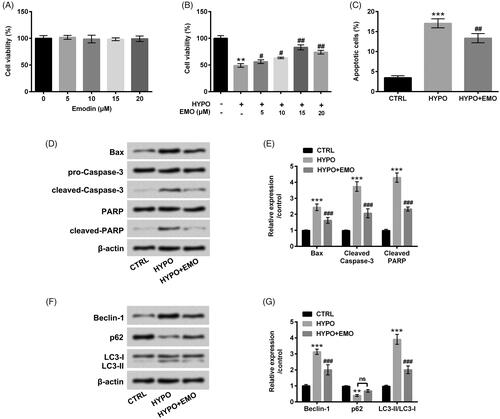
PC-12 cells were next disposed with the disparate concentrations of EMO (5, 10, 15 and 20 μM), concurrently CCK-8 assay was executed for cell cytotoxicity assessment. There was no apparent alteration of PC-12 cell viability after EMO disposition at any concentrations (). However, we discovered that EMO evidently aggrandized cell viability at the concentrations of 5, 10 (p < .05), 15 and 20 μM (p < .01) under hypoxia administration (). EMO at 15 μM was selected as the optimal concentration to dispose PC-12 cells in the follow-up experiments. In , the reduced cell apoptosis (p < .01) and the repressed Bax, cleaved-Caspase-3 and cleaved-PARP expression (p < .001) in hypoxia-disposed PC-12 cells were observed after EMO administration. Furthermore, hypoxia-triggered Beclin-1 and LC3-II enhancements and p62 reduction were both abated by EMO (p < .001, ). These observations hinted that EMO eased hypoxia-triggered PC-12 cells apoptosis and autophagy.
Figure 2. EMO lightened hypoxia-triggered apoptosis and autophagy in PC-12 cells. (A) PC-12 cells were disposed with diverse concentrations of EMO (0, 5, 10, 15 and 20 μM) for 12 h, CCK-8 was implemented for cell cytotoxicity determination. After hypoxia administration and EMO treatment, CCK-8 and flow cytometry were performed for (B) cell viability and (C) cell apoptosis assessment. Western blot assay was implemented for (D and E) Bax, pro/cleaved-Cleaved-3, PARP and cleaved-PARP and (F and G) Beclin-1, p62 and LC3-I/LC3-II protein levels determination. The three independent experiments were conducted. **p < .01 and ***p < .001: HYPO group versus CTRL group; ##p < .01, ###p < .001: HYPO + EMO group versus HYPO group; ns: no significant difference.
EMO elevated miR-25 expression under hypoxia disposition
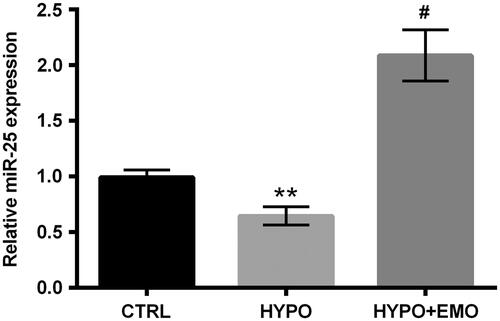
The relative miR-25 expression in PC-12 cells was appraised after disposition with hypoxia and EMO. The repression of miR-25 was discovered in PC-12 cells under hypoxia administration (p < .01). But, EMO disposition dramatically augmented miR-25 expression in hypoxia-stimulated PC-12 cells (p < .05, ). Above findings indicated that EMO could accelerate miR-25 expression in PC-12 cells under hypoxia disposition, hinting that miR-25 might be linked to regulate EMO-affected apoptosis and autophagy in PC-12 cells.
Figure 3. EMO elevated miR-25 expression under hypoxia administration. After hypoxia and EMO (15 μM) disposition, RT-qPCR essay was carried out for miR-25 expression determination in PC-12 cells. The three independent experiments were conducted. **p < .01: HYPO group versus CTRL group; #p < .05: HYPO + EMO group versus HYPO group.
Repression of miR-25 abated the diminished function of EMO in hypoxia-triggered PC-12 cells apoptosis and autophagy
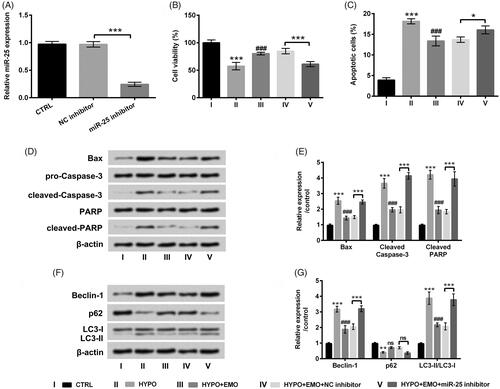
To further verify whether miR-25 is a pivotal mediator in EMO-affected apoptosis and autophagy in PC-12 cells under hypoxia disposition, miR-25 inhibitor was transfected into PC-12 cells to repress miR-25 expression. The result in disclosed that miR-25 expression was evidently restrained in miR-25 inhibitor-transfected PC-12 cells as contrasted with that in the interrelated control group (p < .001). Functions experimental results disclosed that repressed miR-25 significantly hindered cell viability (p < .001) and evoked apoptosis in hypoxia and EMO co-disposed PC-12 cells (p < .05, ). Additionally, miR-25 inhibition also facilitated Bax, cleaved-Caspase-3 and cleaved-PARP expression in hypoxia and EMO co-disposed PC-12 cells (p < .001, ). Beyond that, Beclin-1 and LC3-II expression was elevated but p62 expression was declined by miR-25 repression in PC-12 cells after administration with hypoxia and EMO (p < .001, ). The involved data from above experiments corroborated that suppressed miR-25 could abrogate the impacts of EMO on hypoxia-triggered apoptosis and autophagy in PC-12 cells.
Figure 4. Suppressed miR-25 overturned the impacts of EMO on apoptosis and autophagy in hypoxia-disposed PC-12 cells. (A) After miR-25 inhibitor and its correlative control transfections, RT-qPCR essay was implemented for miR-25 expression detection. PC-12 cells were then co-disposed with hypoxia and EMO, meanwhile transfected with miR-25 inhibitor, CCK-8 and flow cytometry were implemented for (B) cell viability and (C) cell apoptosis detection. Western blot assay was executed for (D and E) Bax, pro/cleaved-Cleaved-3, PARP and cleaved-PARP and (F and G) Beclin-1, p62 and LC3-I/LC3-II protein levels assessment. The three independent experiments were conducted. *p < .05, **p < .01 and ***p < .001: HYPO group versus CTRL group or HYPO + EMO + miR-25 inhibitor versus HYPO + EMO + NC inhibitor; ###p < .001: HYPO + EMO group versus HYPO group; ns: no significant difference.
NEFL was a neoteric target gene of miR-25
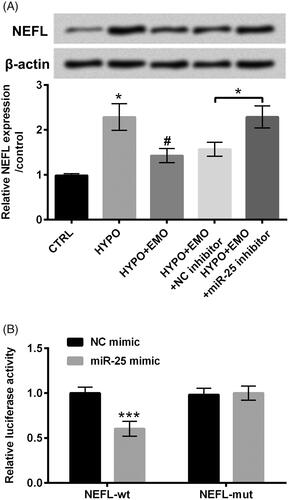
The relevancy between NEFL and miR-25 was subsequently uncovered via estimating the protein and mRNA levels of NEFL in PC-12 cells after disposition with hypoxia and EMO, meanwhile transfection with miR-25 inhibitor. Results in revealed that NEFL protein and mRNA levels were both elevated under hypoxia stimulation (p < .05). After disposition with EMO, the enhancement of NEFL induced by hypoxia was overtly abated (p < .05). But, repression of miR-25 apparently enhanced NEFL expression in hypoxia and EMO co-disposed PC-12 cells (p < .05). These discoveries suggested that EMO hindered NEFL expression via elevation of miR-25. To further probe whether NEFL was a direct target gene of miR-25, dual luciferase report assay was executed. We observed that the luciferase activity was evidently restrained in miR-25 mimic and NEFL-wt co-transfected cells as compared with that in NC mimic and NEFL-wt co-transfected cells (p < .001, ). There was no distinct change in PC-12 cells co-transfection with miR-25 mimic or NC mimic and NEFL-mut (). These correlative results testified that NEFL might be a neoteric target gene of miR-25.
Figure 5. NEFL was a neoteric target gene of miR-25. (A) PC-12 cells were administrated with hypoxia and EMO, simultaneously transfected with miR-25 inhibitor, Western blot and RT-qPCR were executed for assessment of the protein and mRNA levels of NEFL. (B) Dual luciferase report essay was carried out for confirmation of the relevancy between miR-25 and NEFL. The three independent experiments were conducted. *p < .05 and ***p < .001: HYPO group versus CTRL group or HYPO + EMO + miR-25 inhibitor versus HYPO + EMO + NC inhibitor or miR-25 mimic + NEFL-wt versus NC mimic + NEFL-wt; #p < .05: HYPO + EMO group versus HYPO group.
Overexpressed NEFL ascended cell apoptosis and autophagy in EMO and hypoxia-disposed PC-12 cells
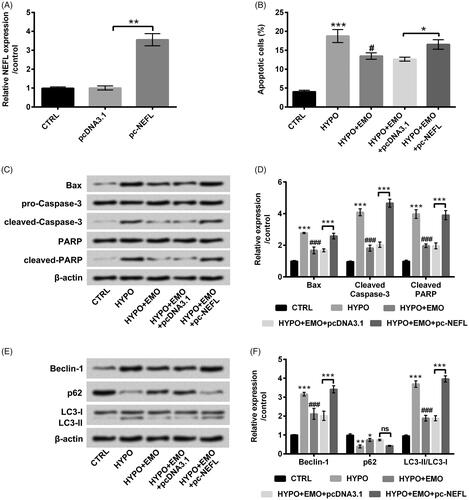
To further probe whether NEFL joined in mediating EMO-affected PC-12 cells apoptosis and autophagy under hypoxia stimulation, the vector of pc-NEFL was transfected into PC-12 cells to overexpress NEFL expression. The result in confirmed the successful transfection (p < .01). Additionally, overexpressed NEFL clearly augmented cell apoptosis (p < .05, ), synchronously elevated Bax, cleaved-Caspase-3 and cleaved-PARP expression in hypoxia and EMO co-administrated PC-12 cells (p < .001, ). Otherwise, overexpressed NEFL notably ascended Beclin-1 and LC3-II protein levels (p < .001) but slightly p62 protein level in hypoxia and EMO co-administrated PC-12 cells (). The above-involved consequences disclosed that overexpressed NEFL could overturn the impeded impacts of EMO on PC-12 cell apoptosis and autophagy under hypoxia disposition.
Figure 6. Overexpressed NEFL ameliorated the functions of EMO in apoptosis and autophagy in hypoxia-disposed PC-12 cells. (A) PC-12 cells were transfected with pc-NEFL and pcDNA3.1 vectors, RT-qPCR essay was implemented for NEFL expression determination in PC-12 cells. After co-disposition with hypoxia and EMO and transfection with pc-NEFL, flow cytomentry was carried out for (B) cell apoptosis assessment. Western blot assay was executed for (C and D) Bax, pro/cleaved-Cleaved-3, PARP and cleaved-PARP and (E and F) Beclin-1, p62 and LC3-I/LC3-II protein levels determination. The three independent experiments were conducted. *p < .05, **p < .01 and ***p < .001: pc-NEFL group versus pcDNA3.1 group, HYPO group versus CTRL group or HYPO + EMO + pc-NEFL versus HYPO + EMO + pcDNA3.1; #p < .05, ###p < .001: HYPO + EMO group versus HYPO group; ns: no significant difference.
EMO activated mTOR and Notch pathways via repression of NEFL in hypoxia-disposed PC-12 cells
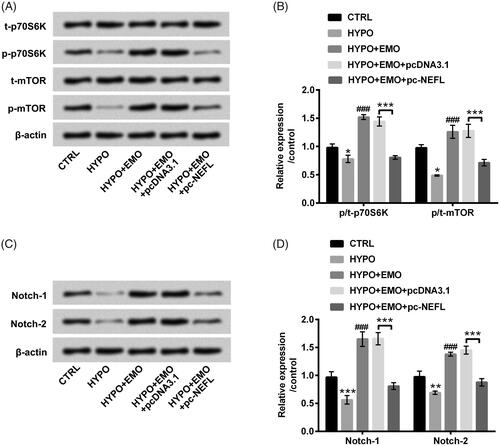
The pivotal signaling pathways of mTOR and Notch were ultimately evaluated to further explore the latent mechanisms. In , results disclosed that hypoxia disposition hindered phosphorylated p70S6K and mTOR expression (p < .05). After administration with EMO, we discovered that hypoxia-triggered suppressive impacts on mTOR signaling pathway were reversed (p < .001). Nonetheless, overexpressed NEFL hindered the activation of mTOR signaling pathway under EMO and hypoxia co-disposition (p < .001). Likewise, Notch-1 and Notch-2 expression levels were also declined by hypoxia administration (p < .01 or p < .001), but elevated by EMO disposition (p < .001). Nevertheless, overexpressed NEFL also impeded Notch signaling pathway in EMO and hypoxia co-stimulated PC-12 cells (p < .001, ). These consequences hinted that EMO could activate mTOR and Notch pathways via restraining NEFL expression in hypoxia-disposed PC-12 cells, which indicated that mTOR and Notch pathways might be momentous mediators in EMO-affected apoptosis and autophagy in PC-12 cells.
Figure 7. EMO activated mTOR and Notch pathways via hindering NEFL expression in hypoxia-disposed PC-12 cells. After co-disposition with hypoxia and EMO and transfection with pc-NEFL, Western blot essay was carried out for (A and B) p/t-p70S6K and p/t-mTOR, as well as (C and D) Notch-1 and Notch-2 protein levels determination. The three independent experiments were conducted. *p < .05, **p < .01 and ***p < .001: HYPO group versus CTRL group or HYPO + EMO + pc-NEFL versus HYPO + EMO + pcDNA3.1; ###p < .001: HYPO + EMO group versus HYPO group.
Discussion
In the actual study, we discovered that EMO evidently mitigated hypoxia-triggered PC-12 cell injury via mediating apoptosis and autophagy. Moreover, EMO augmented miR-25 expression in PC-12 cells under hypoxia disposition. The enhancement of miR-25 might be linked to the protective action of EMO in PC-12 cells. Additionally, we discovered that NEFL might possibly be a neoteric target gene of miR-25, and overexpressed NEFL overturned the protective action of EMO on hypoxia-triggered PC-12 cell injury. Beyond that, we observed that EMO activated mTOR and Notch pathways through hindering NEFL expression in hypoxia-disposed PC-12 cells. These explorations disclosed that EMO might be a valid therapeutic agent for cerebral ischemia.
EMO has been testified to be a neoteric type of anticancer drug with selective activity against neuro-ectodermal cancers [Citation24]. More importantly, a growing number of researches have corroborated that EMO might be a neuroprotective agent for remedy nervous system diseases [Citation17,Citation25]. Cerebral ischemia injury belongs to central nervous system (CNS) injury, which gives rise to irreversible brain damage and even neurological sequelae [Citation26]. Hence, our research attempted to further disclose whether EMO emerges the protective action in cerebral ischemia injury. Apoptosis and autophagy are momentous biological variation in the process of cerebral ischemia injury [Citation27]. PC-12 cells in the actual research were disposed under hypoxia condition to stimulate the process of cerebral ischemia injury. We discovered that hypoxia administration accelerated PC-12 cell apoptosis and autophagy. But, the phenomenon was evidently abated by EMO disposition. The results were partly similar to Chen et al. uncovered that EMO lightened TNF-α-induced apoptosis and autophagy in mouse C2C12 myoblasts [Citation28]. These observations indicated that EMO weakened hypoxia-triggered PC-12 cell injury.
The importance of miRNAs in mediation of the therapeutic activities of EMO has been verified in the existing studies. An interesting research from Xiang et al. attested that EMO could relieved pancreatic acinar cell injury triggered by sodium taurocholate through targeting miR-30a-5p [Citation20]. Otherwise, Hua et al. testified that miR-126 participated in mediating the preventive impacts of EMO on intima thickness in balloon-injured carotid artery rats [Citation29]. MiR-25 is an obbligato miRNA, which has been corroborated to protect PC-12 cells against H2O2-evoked oxidative damage through Wnt/β-catenin pathway [Citation30]. Moreover, miR-25 could repress cerebral ischemia/reperfusion injury-evoked cell apoptosis via mediating Fas/FasL pathway [Citation31]. Nonetheless, whether miR-25 also joins in mediating the protective actions of EMO in hypoxia-triggered PC-12 cell injury remains unreported. The discoveries in our research disclosed that suppressed miR-25 abated the functions of EMO in hypoxia-triggered PC-12 cells, which were indicated that miR-25 might be a pivotal controller in the process of cerebral ischemia injury.
NEFL is located on the short arm of chromosome eight, which genetic variants can affect neuroblastoma susceptibility [Citation32]. As a latent tumour suppresser, NEFL has been testified to mediate cell growth and invasion in head and neck squamous cell carcinoma cells [Citation33]. A crucial research uncovered that NEFL expression was specifically regulated by miR-b1336 and miR-b2403, which might be a direct target gene of the two miRNAs [Citation34]. More interestingly, Peng et al. discovered that miR-25 augmented cell proliferation and invasion in glioblastoma cells via immediately targeting NEFL [Citation35]. Our study disclosed that NEFL expression was repressed by EMO administration, but elevated by miR-25 inhibition in PC-12 cells under hypoxia stimulation, which was also predicated as a latent target gene of miR-25. Outside of this, we revealed that overexpressed NEFL overturned the functions of EMO-affected cell apoptosis and autophagy in hypoxia-injured PC-12 cells. These explorations hinted the involvements of NEFL in regulating the protective action of EMO in hypoxia-injured PC-12 cells.
mTOR and Notch are two momentous signal transduction pathways, which hold the post of regulators in cerebral ischemic injury. An animal experimentation disclosed that repression of mTOR signalling awarded protection against cerebral ischemic injury in acute hyperglycaemic rats [Citation36]. Additionally, activation of Notch pathway has been proven to elevate the microglia-regulated inflammatory response linked to focal cerebral ischemia [Citation37]. Moreover, EMO has been corroborated to restrain TGF-β1-evoked epithelial-mesenchymal transition in alveolar epithelial cells through mediation of Notch pathway [Citation38]. In the actual research, we discovered that EMO enhanced the activations of mTOR and Notch pathways under hypoxia disposition. Nonetheless, the enhancements were evidently reversed by overexpressed NEFL. These observations implied that EMO eased hypoxia-triggered injury possibly mediated by mTOR and Notch pathways. Further researches are still necessary for further certifying the above surmise.
Taken together, the explorations disclosed that EMO conferred protection against hypoxia-triggered injury in PC-12 cells. This phenomenon might be mediated by enhancement of miR-25 via targeting NEFL and via activating mTOR and Notch pathways. These discoveries corroborated that EMO might be a valid remedial agent for cerebral ischemia. The predominant limitation of the research is the lack of in vivo experiment, which will be explored in the future.
Acknowledgements
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Disclosure statement
Authors declare that there is no conflict of interest.
Related Research Data
References
- Brait VH, Arumugam TV, Drummond GR, et al. Importance of T lymphocytes in brain injury, immunodeficiency, and recovery after cerebral ischemia. J Cereb Blood Flow Metab. 2012;32:598–611.
- Kim T, Vemuganti R. Mechanisms of Parkinson’s disease-related proteins in mediating secondary brain damage after cerebral ischemia. J Cereb Blood Flow Metab. 2017;37:1910–1926.
- Vespa P, Bergsneider M, Hattori N, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab. 2005;25:763–774.
- Zhang H, Meng LZ, Lyon R, et al. Monitoring cerebral ischemia during cerebrovascular surgery. J Biomed Res. 2017;31:279–282.
- Park SY, An SA, Lee HB, et al. Different impact of hyperhomocysteinemia on cerebral small vessel ischemia and cervico-cerebral atherosclerosis in non-stroke individuals. Thromb Res. 2013;131:e12–16.
- Schimidt HL, Vieira A, Altermann C, et al. Memory deficits and oxidative stress in cerebral ischemia–reperfusion: neuroprotective role of physical exercise and green tea supplementation. Neurobiol Learn Mem. 2014;114:242–250.
- Wang H, Tri Anggraini F, Chen X, et al. Embryonic lethal abnormal vision proteins and adenine and uridine-rich element mRNAs after global cerebral ischemia and reperfusion in the rat. J Cereb Blood Flow Metab. 2017;37:1494–1507.
- Tian T, Zhang HX, He CP, et al. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials. 2018;150:137–149.
- Sekhon MS, Ainslie PN, Griesdale DE. Clinical pathophysiology of hypoxic ischemic brain injury after cardiac arrest: a “two-hit” model. Crit Care. 2017;21:90.
- Lu Y, Yang JH, Li X, et al. Emodin, a naturally occurring anthraquinone derivative, suppresses IgE-mediated anaphylactic reaction and mast cell activation. Biochem Pharmacol. 2011;82:1700–1708.
- Su J, Yan Y, Qu J, et al. Emodin induces apoptosis of lung cancer cells through ER stress and the TRIB3/NF-κB pathway . Oncol Rep. 2017;37:1565–1572.
- Tian SL, Yang Y, Liu XL, et al. Emodin attenuates bleomycin-induced pulmonary fibrosis via anti-inflammatory and anti-oxidative activities in rats. Med Sci Monit. 2018;24:1–10.
- Janeczko M, Masłyk M, Kubiński K, et al. Emodin, a natural inhibitor of protein kinase CK2, suppresses growth, hyphal development, and biofilm formation of Candida albicans. Yeast. 2017;34:253–265.
- Zhao XA, Chen G, Liu Y, et al. Emodin alleviates liver fibrosis of mice by reducing infiltration of Gr1(hi) monocytes. Evidence-Based Complement Alternat Med. 2018;2018:1–5738101.
- Li L, Sheng X, Zhao S, et al. Nanoparticle-encapsulated emodin decreases diabetic neuropathic pain probably via a mechanism involving P2X3 receptor in the dorsal root ganglia. Purinergic Signal. 2017;13:559–568.
- Li Y, Wang L, Tu Y, et al. A new dosage form of emodin: for solubility and dissolution rate enhancement and application in Alzheimer’s disease and bacteriostasis. J Drug Deliv Sci Technol. 2015;29:261–268.
- Liu T, Jin H, Sun QR, et al. Neuroprotective effects of emodin in rat cortical neurons against beta-amyloid-induced neurotoxicity. Brain Res. 2010;1347:149–160.
- Xu J, Wang Y, Tan X, et al. MicroRNAs in autophagy and their emerging roles in crosstalk with apoptosis. Autophagy. 2012;8:873–882.
- Ouyang YB, Stary CM, Yang GY, et al. microRNAs: innovative targets for cerebral ischemia and stroke. Curr Drug Targets. 2013;14:90–101.
- Xiang H, Tao X, Xia S, et al. Emodin alleviates sodium taurocholate-induced pancreatic acinar cell injury via microRNA-30a-5p-mediated inhibition of high-temperature requirement A/transforming growth factor beta 1 inflammatory dignaling. Front Immunol. 2017;8:1488–1488.
- Pan L, Huang BJ, Ma XE, et al. MiR-25 protects cardiomyocytes against oxidative damage by targeting the mitochondrial calcium uniporter. Int J Mol Sci. 2015;16:5420–5433.
- Liu Q, Wang Y, Yang T, et al. Protective effects of miR-25 against hypoxia/reoxygenation-induced fibrosis and apoptosis of H9c2 cells. Int J Mol Med. 2016;38:1225–1234.
- Weng H, Pan A, Yang L, et al. Estimating number of transgene copies in transgenic rapeseed by real-time PCR assay withHMG I/Y as an endogenous reference gene. Plant Mol Biol Rep. 2004;22:289–300.
- Pecere T, Gazzola MV, Mucignat C, et al. Aloe-emodin is a new type of anticancer agent with selective activity against neuroectodermal tumors. Cancer Res. 2000;60:2800–2804.
- Ho SL, Poon CY, Lin C, et al. Inhibition of beta-amyloid aggregation by albiflorin, aloeemodin and neohesperidin and their neuroprotective effect on primary hippocampal cells against beta-amyloid induced toxicity. Curr Alzheimer Res. 2015;12:424–433.
- Lu X, Gu R, Hu W, et al. Upregulation of heme oxygenase-1 protected against brain damage induced by transient cerebral ischemia-reperfusion injury in rats. Exp Ther Med. 2018;15:4629–4636.
- Shu S, Li CM, You YL, et al. Electroacupuncture ameliorates cerebral ischemia-reperfusion injury by regulation of autophagy and apoptosis. Evid Based Complement Alternat Med. 2016;2016:7297425.
- Chen D, Liu J, Lu L, et al. Emodin attenuates TNF-α-induced apoptosis and autophagy in mouse C2C12 myoblasts though the phosphorylation of Akt. Int Immunopharmacol. 2016;34:107–113.
- Hua JY, He YZ, Xu Y, et al. Emodin prevents intima thickness via Wnt4/Dvl-1/beta-catenin signaling pathway mediated by miR-126 in balloon-injured carotid artery rats. Exp Mol Med. 2015;47:e170.
- Guo Y, Niu S. MiR-25 protects PC-12 cells from H2O2 mediated oxidative damage via WNT/β-catenin pathway . J Spinal Cord Med. 2018;41:416–425.
- Zhang JF, Shi LL, Zhang L, et al. MicroRNA-25 negatively regulates cerebral ischemia/reperfusion injury-induced cell apoptosis through Fas/FasL pathway. J Mol Neurosci. 2016;58:507–516.
- Capasso M, Diskin S, Cimmino F, et al. Common genetic variants in NEFL influence gene expression and neuroblastoma risk. Cancer Res. 2014;74:6913–6924.
- Huang Z, Zhuo Y, Shen Z, et al. The role of NEFL in cell growth and invasion in head and neck squamous cell carcinoma cell lines. J Oral Pathol Med. 2014;43:191–198.
- Ishtiaq M, Campos-Melo D, Volkening K, et al. Analysis of novel NEFL mRNA targeting microRNAs in amyotrophic lateral sclerosis. PLoS One. 2014;9:e85653.
- Peng G, Yuan X, Yuan J, et al. miR-25 promotes glioblastoma cell proliferation and invasion by directly targeting NEFL. Mol Cell Biochem. 2015;409:103–111.
- Fu L, Huang L, Cao C, et al. Inhibition of AMP-activated protein kinase alleviates focal cerebral ischemia injury in mice: interference with mTOR and autophagy. Brain Res. 2016;1650:103–111.
- Wei Z, Chigurupati S, Arumugam TV, et al. Notch activation enhances the microglia-mediated inflammatory response associated with focal cerebral ischemia. Stroke. 2011;42:2589–2594.
- Gao R, Chen R, Cao Y, et al. Emodin suppresses TGF-beta1-induced epithelial-mesenchymal transition in alveolar epithelial cells through Notch signaling pathway. Toxicol Appl Pharmacol. 2017;318:1–7.