
Abstract
Aims
microRNAs (miRNAs) occupy a vital position in diseases. This research aims to detect miR-103 influence on LPS-resulted HK-2 cells inflammation damage.
Methods
Cells were exposed to LPS for inducing injury. CCK-8 and flow cytometry were introduced to detect cell viability and apoptosis rate, respectively. ELISA, qRT-PCR and western blot were respectively used to explore inflammation factors production and relative proteins expression. The relationship between miR-103 and c-Myc was analysed through luciferase reporter assay.
Results
LPS led to significant inhibition of cell viability (p < .05) and proliferation-related proteins expression. It also increased the apoptosis rate (p < .05) and promoted inflammation cytokines overproduction (p < .001). Besides, miR-103 was elevated by LPS (p < .01), and aggravated LPS-induced damage above, while miR-103 inhibitor attenuated this impairment (p < .05, p < .01 or p < .001). However, c-Myc silence abolished miR-103 inhibitor protective effect on cell proliferation (p < .05), apoptosis (p < .01) and inflammation factors expression (p < .05 or p < .01), representing a negative relationship between them. Besides, the activity of NF-κB as well as JAK/STAT pathways was controlled by miR-103, also, mediated through c-Myc.
Conclusions
miR-103 mimic enhanced LPS-caused inflammation damage by silencing c-Myc in HK-2 cell line.
Introduction
Chronic kidney disease (CKD) results from abnormalities in kidney whose structure or function altered irreversibly over 3 months or years [Citation1]. It always has health implications and exhibits with high mortality as well as several clinical symptoms, such as platelet dysfunction, immune dysfunction, increased fractures risk, cardiovascular disease and inflammation response [Citation2,Citation3]. In reality, CKD is characterized as a low-grade inflammation process, and oxidative stress along with inflammation could facilitate this renal function deterioration [Citation4]. Therefore, exploring the regulatory factors and mechanisms of inflammation on the kidney is pivotal for figuring out CKD pathogenesis.
microRNAs (miRNAs) comprise a manifold class of 21–24 nucleotides (nt) noncoding RNAs (ncRNAs) which can regulate the expression of genes by targeting messenger RNAs (mRNAs) [Citation5]. Recent studies have shown that miRNAs are proposed to contribute to CKD or inflammation development. For example, miR-221 and miR-222 performed a relevant function in chronic inflammation by inhibiting endothelial cells (Ecs) proliferation and angiogenesis [Citation6]. miR-103 is diffusely distributed in human tissues, belonging to the miR-103/107 family [Citation7]. It could relate to chronic inflammation caused by Helicobacter pylori and attenuate cardiomyocyte hypertrophy comparatively via cardiac autophagy activity reduction [Citation8]. However, little is known about the role of miR-103 in human proximal tubular cells.
More interesting, in glucocorticoid (GC)-sensitive leukaemia cells, miR-103 was upregulated and reduced c-Myc expression by directing c-Myb, the c-Myc activators [Citation9]. The c-Myc belongs to proto-oncogenic transcription factors family which has been considered involving in tumour improvement [Citation10,Citation11], however, with the advancement of molecular biology, investigations have revealed that c-Myc plays a key role in inflammation regulation. As reported in colitis-associated cancer (CAC), c-Myc specifically correlated with pro-inflammation cytokines production, such as interleukin-1β (IL-1β) and IL-6 by adjusting CUL4A and CUL4B which were specifically overexpressed in CAC tumour via binding to a conserved site located in their promoters [Citation12].
In our experiment, for further discussing the pathological mechanisms of miR-103 and c-Myc in CDK, cells were treated with LPS for the following test, and we explored the roles and relationship of miR-103 as well as c-Myc in LPS-induced inflammation injury, providing a new insight for CDK intervention and cure.
Materials and methods
Cells cultivation and LPS treatment
We took HK-2 cell line (Riken Cell Bank, Ibaraki, Japan) as the material in this study. The RPMI-1640 medium (Wako, Osaka, Japan) containing 100 U/ml penicillin and 0.1 mg/ml streptomycin (Gibco, Carlsbad, CA), respectively, as well as 10% foetal bovine serum (FBS) (Gibco, Carlsbad, CA) was utilized to culture cells, in a 37 °C humidified atmosphere with 5% CO2 and 95% air.
Besides, a series gradient dilutions of LPS (0, 1, 5 and 10 μg/ml) were set up for inducing HK-2 cells inflammation injury through 5 h treatment.
Expression vectors construction and cells transfection
Through GenePharma Co. (Shanghai, China), miR-103 mimic, miR-103 inhibitor, and their negative controls (NCs) were manufactured to alter the expression of miR-103 in HK-2 cells. In addition, si-c-Myc and its NC were used to change c-Myc expression as well. Finally, lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA) was consumed by cells transfection, in line with the direction.
Cell viability assay
Cell viability was identified in this experiment relying on (CCK-8) (Dojindo Molecular Technologies, Gaithersburg, MD). According to the instruction, the CCK-8 solution was added into 96-well plates which contained 5000 cells/well, after a 24 h conventional culture in an environment containing 5% CO2 along with 95% air at 37 °C, and CCK-8 incubated with the cells for another 1 h under same conditions. After that, a microplate reader (Bio-Rad, Hercules, CA) was introduced for absorbance measurement under 450 nm.
Cell apoptosis assay
The cells apoptotic rate was measured by Annexin V-FITC/PI apoptosis detection kit (Beijing Biosea Biotechnology, Beijing, China) combined with flow cytometry (Beckman Coulter, Brea, CA). First, cells were adjusted and seeded in the six-well plate with 1 × 105 cells/well density and cultured at 37 °C for 24 h. Then, these treated cells were washed and resuspended with 4 °C phosphate-buffered saline (PBS); 5 μl Annexin V-FITC and 5 μl PI were mixed with the cell suspension in the dark for 15 min to constitute the sample for detecting flow cytometry .
Western blot
After cells collection, 1 × PBS buffer and 4 °C RIA lysis buffer (Beyotime Biotechnology, Shanghai, China) supplemented with protease inhibitors (Roche, Guangzhou, China) were respectively used for rinse and proteins extraction. The BCA™ Protein Assay Kit (Pierce, Appleton, WI) was applied to proteins quantification and the Bio-Rad Bis-Tris Gel system (Bio-Rad, Shanghai, China) was helped to establish the western blot system. Primary antibodies were diluted using 5% blocking buffer at 1:1000, and incubated with the polyvinylidene difluoride (PVDF) membrane, which carried proteins blots overnight at 4 °C and washed by tris-buffered saline and tween 20 (TBST). The primary antibodies include anti-proliferating cell nuclear antigen (PCNA) (ab92552), anti-Cyclin A2 (ab211735), anti-Cyclin E1 (ab33911), anti-Cyclin-dependent kinase 2 (CDK2) (ab32147), anti-Cyclin D1 (ab40754), anti-CDK4 (ab108357), anti-Bcl-2 (ab32124), anti-Bax (ab32503), anti-pro-caspase-3 (ab32150), anti-cleaved-caspase-3 (ab208003), anti-pro-caspase-9 (ab138412), anti-cleaved-caspase-9 (ab2324), anti-c-Myc (ab32072), anti-phosphorylated Janus kinase (p-Jak1) (ab138005), anti-Jak1 (ab138005), anti-Tyk2 (ab138394), anti-tyrosine kinase 2 (Tyk2), (ab138394), anti-p-signal transducers and activators of transcription 1 (stat1) (ab30645), anti-stat1 (ab3987), anti-p-stat2 (ab53132), anti-stat2 (ab239811), anti-p-p65 (ab28856), anti-p65 (ab32536), anti-inhibitor of nuclear factor κB (NF-κB) kinase α (IκBα) (ab32518), anti-p-IκB α (ab24783) and anti-GAPDH (ab22555). Following 1 h attachment of horseradish peroxidase signed secondary antibody, at room temperature, the PVDF membrane was transferred into the Bio-Rad ChemiDoc™ XRS system (Bio-Rad, Shanghai, China) after 200 μl Immobilon Western Chemiluminescent Horseradish Peroxidase (HRP) Substrate (Millipore, Billerica, MA) was added. Image Lab™ Software (Bio-Rad, Shanghai, China) was adopted to capture and quantify the protein bands.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Both HK-2 cells disruption and total RNA extraction were carried out through Trizol reagent (Life Technologies Corporation, Carlsbad, CA) in keeping with the manufacturer’s description. Taqman MicroRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) was used for total RNA reversing; and Taqman Universal Master Mix II with the TaqMan MicroRNA Assay (Applied Biosystems, Foster City, CA) was used for qRT-PCR. Experiment records were managed through 2–ΔΔCt method and normalized with U6 or GAPDH.
Enzyme-linked immunosorbent assay (ELISA)
After treatments, the culture supernatant of HK-2 cells was collected from 24-well plates and the relative inflammation cytokines, consisting of IL-1β, IL-6, IL-8 and tumour necrosis factor-α (TNF-α) production measured by specific ELISA kits (R&D Systems, Abingdon, UK).
Luciferase reporter assay
The 3′ untranslated regions (UTRs) carrying miR-103-binding site were generated by PCR, and then it was cloned into pmiR-report vector (Genepharma, Shanghai, China) to form the c-Myc-wild-type report vector (c-Myc-wt) and the c-Myc-mutant-type (c-Myc-mt) report vector whose miR-103-binding site was replaced. miR-103 mimic or scramble with the c-Myc-wt or the c-Myc-mt was co-transfected into HEK293T cells using lipofectamine 3000 (Thermo Fisher, Waltham, MA) and reporter assays were performed depending on the dual-luciferase assay system (Promega, Madison, WI) following the manufacturer’s information.
Statistical analysis
Every experiment was replayed at least three times, and exhibited as mean ± SD. SPSS 19.0 statistical software (IBM, Armonk, NY) was applied to statistical analysis. p Values were calculated using a one-way analysis of variance (ANOVA) or t-test and p< .05 was considered a statistically significant result.
Results
LPS induces inflammation injury on HK-2 cells
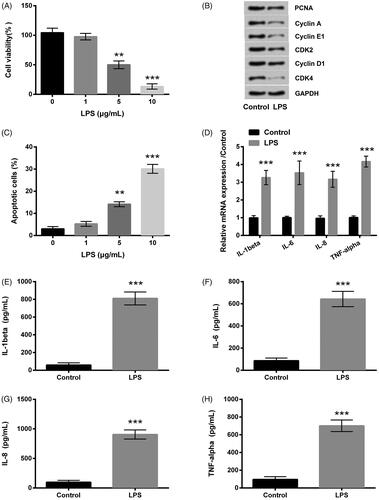
HK-2 cells were handled with series gradient concentrations of LPS solutions, including 0, 1, 5 and 10 μg/ml. Results indicated that in contrast to the control group, cell viability (p < .01 or p < .001, ) as well as PCNA, CyclinA, CyclinE, CyclinD1, CDK2 and CDK4 proteins expression levels () were significantly decreased by LPS of 5 or 10 μg/ml; however, cell apoptosis rate was markedly elevated (p < .01 or p < .001, ). All changes were in a dose-reliant manner; therefore, 5 μg/ml LPS was picked out for the following test. qRT-PCR showed that IL-1β, IL-6, IL-8 and TNF-α mRNA expression levels were apparently increased (all p < .001, ). ELISA implied that the productions of IL-1β, IL-6, IL-8 and TNF-α were all drastically improved (all p < .001, ).
Figure 1. LPS prompted inflammation impairment in HK-2 cells. (A–C) The cell viability, cycle-related proteins expression and the apoptotic rate were detected by CCK-8, western blot and Annexin V-FITC/PI kit combined with flow cytometry. (D) qRT-PCR showed that the inflammatory factors mRNA expression levels were promoted by LPS. (E–H) The inflammatory factors were determined by ELISA. LPS: lipopolysaccharide; CCK-8: cell counting kit-8; FITC: fluorescein isothiocyanate; PI: propidium iodide; qRT-PCR: quantitative real-time polymerase chain reaction; mRNA: messenger RNA; ELISA: enzyme-linked immunosorbent assay. **p < .01, ***p < .001 compared to the corresponding group.
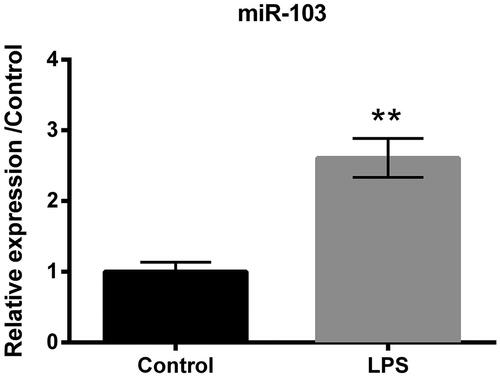
LPS up-regulates the expression of miR-103
qRT-PCR was conducted to verify the miR-103 expression level after the treatment of 5 μg/ml LPS in HK-2 cells. Results hinted that miR-103 expression was highly promoted by LPS (p < .01, ), compared to the control group. It was clear that miR-103 was related to LPS-induced inflammation damage in HK-2 cell line.
Figure 2. LPS led to miR-103 overproduction. The expression level of miR-103 was elevated by LPS. LPS: lipopolysaccharide; miR-103: microRNA-103. **p < .01 compared to the corresponding group.
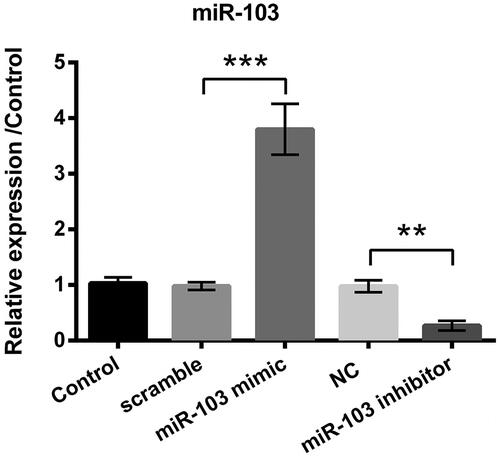
miR-103 mimic and miR-103 inhibitor alters miR-103 expression level
Compared to the corresponding group, miR-103 mimic statistically improved the relative expression level of miR-103 (p < .001, ) while miR-103 inhibitor played an opposite effect (p < .001, ). At the same time, the scramble and NC vectors did not make any sense compared to the control group (). All data explained that the miR-103 mimic and inhibitor vectors have been successfully constructed and transfected into cells.
Figure 3. miR-103 expression level was altered by miR-103 mimic and miR-103 inhibitor. Outcomes showed that miR-103 overexpressed after the transfection of miR-103 mimic, and miR-103 was decreased after the transfection of miR-103 inhibitor. miR-103 microRNA-103; **p < .01, ***p < .001 compared to the corresponding group.
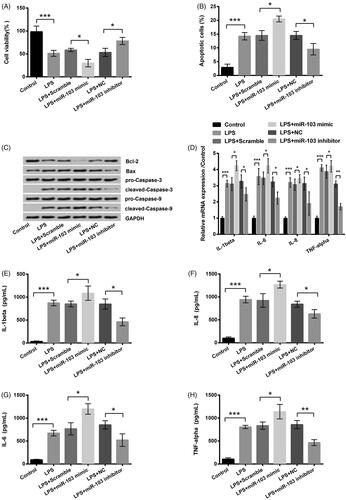
miR-103 aggravates LPS-produced inflammation damage in HK-2 cells
According to the study information, in LPS + miR-103 mimic group, the miR-103 mimic notably enhanced LPS-induced viability inhibition (p < .05, ). Moreover, miR-103 mimic promoted apoptosis, leading to higher apoptosis rate (p < .05, ) and apoptosis-related protein expression levels, excluding Bcl-2 (). Furthermore, qRT-PCR and ELISA exhibited that the IL-1β, IL-6, IL-8 and TNF-α expressions were further promoted by miR-103 mimic (all p < .01, ). In addition, the miR-103 inhibitor exhibited a contrary effect. Compared to the LPS treatment group, scramble and NC had no sense. This phenomenon indicated that miR-103 might act as a proinflammation factor when HK-2 cells underwent 5 h LPS treatment. miR-103 overexpression could aggravate LPS-induced inflammation injury and miR-103 silence had the opposite effect.
Figure 4. Influence of miR-103 on LPS-induced inflammatory injury. miR-103 mimic, miR-103 inhibitor and their negative controls (scramble and NC) were transfected into cells. (A–C) The cell viability, apoptotic rate and the apoptosis-related proteins expression were measured through CCK-8, and Annexin V-FITC/PI kit combined with flow cytometry and western blot, in proper sequence. (D) The inflammatory factors mRNA expression levels were tested by qRT-PCR. (E–H) Inflammatory factors production was determined by ELISA. LPS: lipopolysaccharide; miR-103: microRNA-103; NC: negative control; mRNA: messenger RNA; qRT-PCR: quantitative real-time polymerase chain reaction; CCK-8: cell counting kit-8; FITC: fluorescein isothiocyanate; ELISA: enzyme-linked immunosorbent assay. *p < .05, **p < .01, ***p < .001 in contrast with the corresponding group.
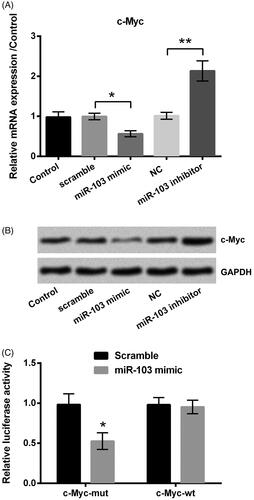
miR-103 targets and down-regulates c-Myc
qRT-PCR as well as western blot was used to identify the expression of c-Myc. Results implied that miR-103 mimic significantly inhibited c-Myc expression at both mRNA level (p < .05, ) and protein level (), compared to the scramble group; however, in contrast with the NC group, c-Myc was up-regulated by the miR-103 inhibitor at the two levels (p < .01, ). What is more, co-transfection of miR-103 mimic and c-Myc-wt led to an obvious lower luciferase activity than that in the miR-103 mimic + c-Myc-mt group (p < .05, ) even though miR-103 scramble did not work significantly. Above all, these data revealed that c-Myc was a target of miR-103, and was negatively controlled by miR-103.
Figure 5. The c-Myc was a target of miR-103. The effect of miR-103 on c-Myc expression was detected by (A) qRT-PCR at the RNA level and (B) western blot at the protein level. (C) The luciferase activity was measured by luciferase reporter assay after the co-transfection of c-Myc-wt or c-Myc-mt and the miR-103 mimic. miR-103: microRNA-103; qRT-PCR: quantitative real-time polymerase chain reaction; c-Myc-wt: c-Myc-wild type; c-Myc-mt: c-Myc-mutant. *p < .05, **p < .01 in contrast with the corresponding group.
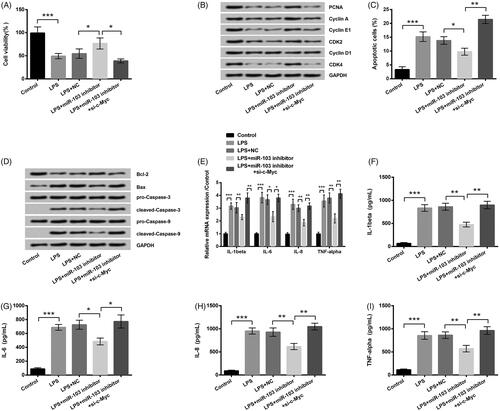
miR-103 inhibitor attenuates LPS-induced inflammation injury by up-regulating c-Myc
For investigating the relationship between the miR-103 and c-Myc, additionally, we examined the proliferation and apoptosis of HK-2 cells. Detection findings showed that miR-103 inhibitor notably alleviated LPS-resulted inflammation injury, but si-c-Myc disturbed this anti-inflammation effect. The cell viability (p < .05, ) and PCNA, CyclinA, CyclinE, CyclinD1, CDK2 and CDK4 proteins levels () were significantly down-regulated; moreover, the apoptosis rate (p < .01, ) and the expression levels of apoptosis-related proteins consisting of Bax, cleaved-caspase 3 and cleaved-caspase 9 were highly improved excepting the Bcl-2 (). What is more, the mRNA expression levels as well as the production of the IL-1β (both p < .01), IL-6 (both p < .05), IL-8 (both p < .01) and TNF-α (both p < .01) were evidently up-regulated by si-c-Myc (). This experiment suggested that miR-103 relieved LPS-induced injury in HK-2 cells, which was mediated by c-Myc.
Figure 6. The impact of c-Myc on the protective effect of miR-103 inhibitor. After the respective transfection of miR-103 inhibitor and its negative control (NC) as well as the co-transfection of miR-103 inhibitor and si-c-Myc before LPS treatment, (A) the cell viability, (B) the cycle-related proteins expression, (C) the apoptosis rate, (D) the apoptosis-related proteins, (E) the inflammatory factors mRNA expression level and (F–I) the inflammatory factors real production were assessed. miR-103: microRNA-103; LPS: lipopolysaccharide; mRNA: messenger RNA. *p < .05, **p < .01, ***p < .001 in contrast with the corresponding group.
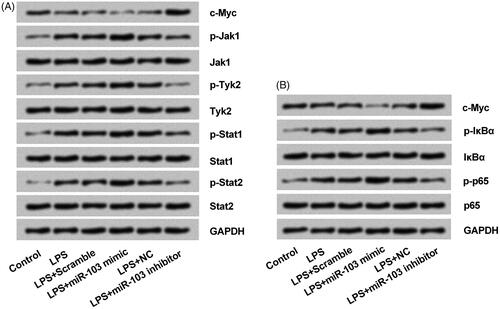
miR-103 regulates NF-κB and JAK/STAT activity by modulating c-Myc
Western blot was conducted to determine whether miR-103-c-Myc axis participated in the NF-κB and JAK/STAT activity. Results implied that LPS markedly decreased c-Myc production but had an opposite effect on the key proteins of NF-κB and JAK/STAT signalling pathways. Furthermore, the c-Myc expression was negatively controlled by the miR-103 (); moreover, p-Jak1, p-Tyk2, p-Stat1, p-Stat2 p-IκBα and p-p65 proteins expression were all facilitated by miR-103 mimic and reduced by miR-103 inhibitor; even there were no statistically changes on Jak1, Tyk2, Stat1 Stat2, IκBα and p65 proteins (). These outcomes displayed that miR-103-c-Myc axis modulated the sensitivity of NF-κB and JAK/STAT signalling pathways. In detail, miR-103 overexpression activated NF-κB and JAK/STAT through c-Myc silence; and miR-103 silence inactivated them by up-regulating c-Myc.
Figure 7. miR-103 regulated the NF-κB and JAK/STAT pathways activity through c-Myc modulation. (A) The expression of phosphorylated key proteins of NF-κB pathway and (B) JAK-STAT pathway were appraised by western blot. NF-κB: nuclear factor κB; JAK: janus kinase; STAT: signal transducers and activators of transcription; miR-103: microRNA-103.
Discussion
To search for the potential CKD pathological mechanisms, here, we took HK-2 cell line as the main study material. After a series of management, including cells transfection, we observed that miR-103 represented a pro-inflammation factor in HK-2 cells. miR-103 mimic facilitated LPS-induced cell proliferation inhibition, apoptosis, as well as inflammation cytokines overproduction by down-regulating c-Myc. Meanwhile, on the contrary, miR-103 inhibitor protected cells from this injury.
The main function of miRNAs is to regulate genes expression by regulating translation or target mRNAs [Citation13]. miRNAs have been recognized as regulators of inflammation responses or CDK owing to the multiplying effects on proliferation, apoptosis, autophagy as well as inflammation cytokines production [Citation14,Citation15]. For example, miR-146a expression was positively associated with the development of CKD [Citation16]. What is more, miR-223-3p and miR-93-5p were reported to show a significant down-regulation in CKD, leading to a stable elevation of IL-6 and IL-8 [Citation17]. miR-103 is geared to the miRNAs family, so it probably manifests similarities with the others. Natarelli et al. found that miR-103 impaired Ecs proliferation by inhibiting Notch homolog 1 (Notch1) activity [Citation18]. Overexpression of miR-103 inhibited the expression of CDK2 as well as its Cyclin E1 target, leading to cell proliferation restraint [Citation9]. In addition, miR-103 showed a stable increase in expression accompanies with high quantity of TNF-α, IL-2 and TGF-β-RII proteins after bacterium eradication treatment in inflammation cells [Citation8]. However, the potential role of miR-103 in CKD is still uncertain.
To address the effects of miR-103, we performed in vivo experiment. LPS was used to mimic inflammatory injury of CKD in HK-2 cells. It was identified that LPS triggered inflammation injury in HK-2 cells in a dose-dependent way and also accelerated an overproduction of miR-103. In addition, we found that miR-103 could aggravate the inflammation damage via causing LPS-induced proliferation inhibition, apoptosis, and of IL-1β, IL-6, IL-8, as well as TNF-α overproduction in turn. The viability, apoptosis rate, the protein expression levels of Bax, cleaved-caspase 3, cleaved-caspase 9 but Bcl-2, as well as the cytokines production, not only the RNA expression level but also the protein level, were visually elevated. On the contrary, miR-103 inhibitor protected HK-2 cells from LPS impairment. All these in vitro study results presented a new opinion and suggested that miR-103 was closely associated with LPS-resulted inflammation injury, and it was harmful to cells survival by acting as a pro-inflammation element.
Previous studies have shown that c-Myc plays a key role in promoting proliferation, self-renewal and survival of cells [Citation19,Citation20]. Moreover, there was a crosstalk between miRNAs and the c-Myc through forming the functional loop [Citation21]. This connection has been indicated in hepatocellular carcinoma cells (HCCs) [Citation22], human macrophages [Citation23] and human nasopharyngeal carcinoma [Citation24] and so on. But, it is rarely discussed in HK-2 cells. Thus, to confirm our conclusion, a further detection was conducted by the dual luciferase activity assay, qRT-PCR and western blot in our research. Out of question, the protective influence of miR-103 inhibitor on HK-2 cells was weakened by si-c-Myc, by comparison with a lower viability and PCNA, CyclinA, CyclinE, CyclinD1, CDK2 also CDK4 proteins levels, but a higher apoptosis rate, and Bax, cleaved-caspase 3 also cleaved-caspase 9 levels, excluding Bcl-2, even more inflammation factors expression massive. Our data further uncovered that c-Myc was a downstream target of miR-103 and c-Myc mediated the effect of miR-103 during LPS treatment, providing an compelling evidence for the NC relationship between them, as well as the anti-inflammation function of c-Myc. In other words, miR-103 inhibitor attenuates LPS-induced inflammation injury by up-regulating c-Myc. Same phenomenon was also observed by Zhao team who established the miR-184 mimic and inhibitor, and reported that miR-184 inhibited colon cancer cells proliferation and promoted the apoptosis by down-regulating c-Myc [Citation25]. In another research, miR-665 suppressed neuroblastoma growth by inhibiting c-Myc and HDAC8 expression [Citation26]. They were consistent with the conclusions derived from our findings.
NF-κB and JAK-STAT are major pathways involving cell cycle progression, mitotic cell division, proliferation, apoptosis and cell communication [Citation27]. Furthermore, it is well known that the NF-κB transcription factor family is activated by pro-inflammation factors and exerts a strong pro-inflammation signal to induce the expression of inflammation cytokines and chemokines [Citation28]. Besides, Wang et al. implied that JAK/STAT pathway is a critical mediator to induce inflammation process and apoptosis [Citation29]. miRNAs function through NF-κB and JAK/STAT pathways. For example, Bardoxolone methyl suppressed pro-inflammation signalling to reduce oxidative stress and inflammation through activating nuclear factor erythroid-derived 2-related factor 2 (Nrf2) and inhibiting NF-κB in CKD [Citation30]. Cruz et al. demonstrated that miRNAs stimulated the secretion of TNF-α mediating by the activation of NF-κB pathway subunit p65 [Citation31]. Through the Toll-like receptor (TLR)/NF-κB pathway, miR-181a dependently involved in the progression of CKD [Citation32]. miR-146b derived from the stimulatory action of inflammation cytokines TNF-α and IL-6, and in turn miR-146b regulated the inflammation process by influencing the NF-κB signalling pathway [Citation31]. However, the relationship between the miR-103, c-Myc and the NF-κB and JAK-STAT pathways in the HK-2 cell line is still waiting for discovering. In our study, miR-103 overexpression stimulated NF-κB and JAK-STAT pathways activation by down-regulating c-Myc that was attached by the low phosphorylation of relative proteins. Vice versa, both pathways’ activity faded with the boost of c-Myc which was caused by the miR-103 inhibitor.
Above all, our achievement found that miR-103 promoted LPS-resulted inflammation injury through NF-κB and JAK-STAT pathways by adjusting c-Myc. It brought a new insight into CKD pathological mechanism and provided a theoretical basis for the treatment of this disease.
Disclosure statement
The authors declare that there are no conflicts of interest.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
- Webster AC, Nagler EV, Morton RL, et al. Chronic kidney disease. Lancet. 2017;389:1238–1252.
- Anders HJ, Andersen K, Stecher B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013;83:1010–1016.
- Ketteler M, Block GA, Evenepoel P, et al. Diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder: synopsis of the kidney disease: improving global outcomes 2017 clinical practice guideline update. Ann Intern Med. 2018;168:422–430.
- Tucker PS, Scanlan AT, Dalbo VJ. Chronic kidney disease influences multiple systems: describing the relationship between oxidative stress, inflammation, kidney damage, and concomitant disease. Oxid Med Cell Longev. 2015;2015:1.
- Yamamura S, Imai-Sumida M, Tanaka Y, et al. Interaction and cross-talk between non-coding RNAs. Cell Mol Life Sci. 2018;75:467–484.
- Hulsmans M, De Keyzer D, Holvoet P. MicroRNAs regulating oxidative stress and inflammation in relation to obesity and atherosclerosis. FASEB J. 2011;25:2515–2527.
- Ellis KL, Cameron VA, Troughton RW, et al. Circulating microRNAs as candidate markers to distinguish heart failure in breathless patients. Eur J Heart Fail. 2013;15:1138–1147.
- Rossi AFT, Cadamuro ACT, Biselli‐Périco JM, et al. Interaction between inflammatory mediators and miRNAs in Helicobacter pylori infection. Cell Microbiol. 2016;18:1444–1458.
- Kfir-Erenfeld S, Haggiag N, Biton M, et al. miR-103 inhibits proliferation and sensitizes hemopoietic tumor cells for glucocorticoid-induced apoptosis. Oncotarget. 2017;8:472.
- Porro A, Haber M, Diolaiti D, et al. Direct and coordinate regulation of ATP-binding cassette transporter genes by Myc factors generates specific transcription signatures that significantly affect the chemoresistance phenotype of cancer cells. J Biol Chem. 2010;285:19532–19543.
- Yap CS, Peterson AL, Castellani G, et al. Kinetic profiling of the c-Myc transcriptome and bioinformatic analysis of repressed gene promoters. Cell Cycle (Georgetown, TX). 2011;10:2184–2196.
- Liu H, Lu W, He H, et al. Inflammation-dependent overexpression of c-Myc enhances CRL4DCAF4 E3 ligase activity and promotes ubiquitination of ST7 in colitis-associated cancer. J Pathol. 2019.
- Rong D, Sun H, Li Z, et al. An emerging function of circRNA-miRNAs-mRNA axis in human diseases. Oncotarget. 2017;8:73271–73281.
- Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15:321.
- Lorenzen JM, Haller H, Thum T. MicroRNAs as mediators and therapeutic targets in chronic kidney disease. Nat Rev Nephrol. 2011;7:286–294.
- Ichii O, Otsuka S, Sasaki N, et al. Altered expression of microRNA miR-146a correlates with the development of chronic renal inflammation. Kidney Int. 2012;81:280–292.
- Ulbing M, Kirsch AH, Leber B, et al. MicroRNAs 223-3p and 93-5p in patients with chronic kidney disease before and after renal transplantation. Bone. 2017;95:115–123.
- Natarelli L, Hartmann P, Wei Y, et al. MiR-103 target lncWDR59 to affect endothelial proliferation balanced by Notch1 and Wnt signaling co-activation. Atherosclerosis. 2017;263:e5.
- Yoshida GJ, Saya H. Inversed relationship between CD44 variant and c-Myc due to oxidative stress-induced canonical Wnt activation. Biochem Biophys Res Commun. 2014;443:622–627.
- Li H, Fang Y, Niu C, et al. Inhibition of cIAP1 as a strategy for targeting c-MYC-driven oncogenic activity. Proc Natl Acad Sci USA. 2018;115:E9317–E9324.
- Jackstadt R, Hermeking H. MicroRNAs as regulators and mediators of c-MYC function. Biochim Biophys Acta. 2015;1849:544–553.
- Bai HY, Liao YJ, Cai MY, et al. Eukaryotic initiation factor 5A2 contributes to the maintenance of CD133 (+) hepatocellular carcinoma cells via the c-Myc/microRNA-29b axis. Stem Cells. 2018;36:180–191.
- Colineau L, Lambertz U, Fornes O. c-Myc is a novel Leishmania virulence factor by proxy that targets the host miRNA system and is essential for survival in human macrophages. J Biol Chem. 2018;293:12805–12819.
- Li M, Liu Y, Wei Y, et al. Zinc-finger protein YY1 suppresses tumor growth of human nasopharyngeal carcinoma by inactivating c-Myc-mediated microRNA-141 transcription. J Biol Chem. 2019;294:6172–6187.
- Wang YB, Zhao XH, Li G, et al. MicroRNA-184 inhibits proliferation and promotes apoptosis of human colon cancer SW480 and HCT116 cells by downregulating C-MYC and BCL-2. J Cell Biochem. 2018;119:1702–1715.
- Prashad N. miR-665 targets c-MYC and HDAC8 to inhibit murine neuroblastoma cell growth. Oncotarget. 2018;9:33186–33201.
- Nandi S, Salman M, Sharma A, et al. Signal transduction through JAK-STAT and NF-κB regulated Pim-1 kinase: novel target for anticancer leads. Curr Signal Transduct. 2018;13:83–104.
- Wu Z, Lu H, Sheng J, et al. Inductive microRNA-21 impairs anti-mycobacterial responses by targeting IL-12 and Bcl-2. FEBS Lett. 2012;586:2459–2467.
- Wang X, Liu Q, Ihsan A, et al. JAK/STAT pathway plays a critical role in the proinflammatory gene expression and apoptosis of RAW264.7 cells induced by trichothecenes as DON and T-2 toxin. Toxicol Sci. 2012;127:412–424.
- Chin MP, Bakris GL, Block GA, et al. Bardoxolone methyl improves kidney function in patients with chronic kidney disease stage 4 and type 2 diabetes: post-hoc analyses from bardoxolone methyl evaluation in patients with chronic kidney disease and type 2 diabetes study. Am J Nephrol. 2018;47:40–47.
- Cruz KJC, de Oliveira ARS, Morais JBS, et al. Role of microRNAs on adipogenesis, chronic low-grade inflammation, and insulin resistance in obesity. Nutrition (Burbank, Los Angeles County, CA). 2017;35:28–35.
- Liu L, Pang XL, Shang WJ, et al. Over-expressed microRNA-181a reduces glomerular sclerosis and renal tubular epithelial injury in rats with chronic kidney disease via down-regulation of the TLR/NF-kappaB pathway by binding to CRY1. Mol Med. 2018;24:49.