
Abstract
JHDM1A participates in cancer development via demethylate dimethyl histone H3 lysine 36 (H3K36me2). p300 is an intrinsic acetyltransferase. This study explored the acetyltransferase activity of p300 on JHDM1A and analyzed the JHDM1A acetylation on H3K36me2 demethylation in osteosarcoma. Co-immunoprecipitation (CoIP) and immunoblotting assay found that p300 directly acetylated JHDM1A at K409 residue in osteosarcoma MG-63 and HOS cells. Nucleosomes and mononucleosomes were prepared and found that acetylation of JHDMIA disrupted its association with nucleosomes and thereby impaired its capability to induce H3K36me2 demethylation. Moreover, chromatin immunoprecipitation (ChIP) assay discovered that the input levels of H3K36me2 in the promoter regions of p21 and puma were increased after acetylation of JHDM1A, which raised the p21 and puma mRNA levels in the cells. Finally, the analysis of JHDM1A acetylation on osteosarcoma cell proliferation and invasion, along with tumor growth pointed out that acetylation of JHDMIA inhibited the proliferation and invasion of osteosarcoma HOS cells, as well as suppressed the tumor growth of osteosarcoma. In conclusion, the outcomes of our research verified that p300 could directly acetylate JHDM1A at K409 site, which reduces the demethylation of H3K36me2, enhanced the transcription of p21 and puma, and thereby inhibited the growth and metastasis of osteosarcoma.
Introduction
Osteosarcoma is the most common malignant bone tumor in children and adolescents [Citation1]. Histone modification is one of the most important fundamental process of epigenetic changes, which includes methylation, acetylation, phosphorylation, ubiquitination and ADP-ribosylation [Citation2]. Aberrant histone modification has been reported to be associated with the occurrence and development of osteosarcoma. [Citation3,Citation4]. It is proved that the posttranslational modification of histones can affect chromatin accessibility, thereby influencing the transcription of a number of genes and multiple cellular biological processes [Citation5,Citation6]. Considering that the 5-year survival rate of non-metastasis osteosarcoma is 60–70% and the 5-year survival rate of metastasis osteosarcoma is less than 20% [Citation1,Citation7], we believe that a better comprehension of histone modification regulation in osteosarcoma cells is of great important sense for developing a novel therapeutic strategy for osteosarcoma.
JHDM1A (also called KDM2A or FBXL11) is a member of JmjC-domain-containing histone demethylase family [Citation8]. Previous studies report that JHDM1A participates in the regulation of gene silencing, cell proliferation and cancer development via demethylate dimethyl histone H3 lysine 36 (H3K36me2) [Citation9,Citation10]. Histone methylation, including H3K36me2, has been demonstrated to exert key roles in the maintenance of genomic stability and the regulation of DNA repair [Citation11]. Histone demethylation, including H3K36me2 demethylation, has been demonstrated to play critical roles in oncogenic reprogramming and cell proliferation regulation [Citation12,Citation13]. The imbalance between histone methylation and demethylation has been found to be associated with cancer tumorigenesis [Citation14]. Wagner et al. indicated that JHDM1A could promote non-small cell lung cancer growth and metastasis by epigenetically activating ERK1/2 signaling [Citation15]. Lu et al. reported that JHDM1A could promote the ovarian cancer development through modulating PI3K pathway and lowering epithelial-mesenchymal transition (EMT) process [Citation16]. Whether JHDM1A participates in the occurrence and development of osteosarcoma is still needed to be explored.
p300 is a central regulator of gene expression in metazoan cells, which presents an intrinsic acetyltransferase activity and can acetylate histones and non-histone proteins at many sites [Citation17,Citation18]. It is reported that there are over 70 proteins that can be acetylated by p300 in cells [Citation18]. However, no literature is available about the acetyltransferase effects of p300 on JHDM1A. More experimental researches are needed.
Therefore, in the current research, we investigated the possible acetyltransferase activity of p300 on JHDM1A and explored the effects of JHDM1A acetylation on H3K36me2 demethylation in osteosarcoma cells. Moreover, the influences of JHDM1A acetylation on osteosarcoma cell growth and metastasis were also analyzed. The findings of our research will be helpful for comprehending the regulatory roles of p300 and JHDM1A in osteosarcoma occurrence and development.
Materials and methods
Plasmids, shRNA, mutagenesis and antibodies
The full-length sequence of wild type (WT) JHDM1A and p300 were expanded using PCR and constructed into Flag-, HA-, His- or Myc-tagged plasmids (TaKaRa Biotechnology, Dalian, China), respectively. The empty plasmid acted as a negative control. Lysine residues 409 (K409) of JHDM1A was substituted with arginine (R) or glutamine (Q) using site-directed mutagenesis. Short-hairpin RNA targeting p300 was ligated into pLKO.1- TRC cloning vector (Addgene, Watertown, MA, USA) to form sh-p300. The nucleotide sequences of sh-p300 were: 5′-GGUUGUUAAUGGAGUUGAA-3′ (sense) and 5′-UUCAACUCCAUUAACAACC-3′ (anti-sense). Following infection, cells were selected by puromycin (Gibco, Carlsbad, CA, USA). Anti-acetyl-lysine antibody (Ac-K, ab21623), anti-Flag antibody (ab49763), anti-JHDM1A antibody (ab174693), anti-p300 antibody (ab54984), anti-Myc antibody (ab9106), anti-HA antibody (ab1424), anti-H3K36me2 antibody (ab9049) and anti-H3K36me3 antibody (ab9050) were all obtained from Abcam Biotechnology (Cambridge, MA, USA).
Cell lines
MG-63, HOS and HEK293 cells were all provided by Cell Bank of Chinese Academy of Science (Shanghai, China) and grown in MEM medium (Gibco) containing 10% fatal bovine serum (FBS, Gibco) and 1% penicillin-streptomycin solution (Gibco) at 37 °C with 5% CO2.
Trichostatin A (TSA) and nicotinamide (NAM) were purchased from Sigma-Aldrich (St. Louis, MO, USA). MG-63 and HOS cells were treated by 100 nM TSA and/or 5 mM NAM in this research.
Co-immunoprecipitation (CoIP) and chromatin immunoprecipitation (ChIP)
CoIP and ChIP were down as previously described [Citation19]. Briefly, cells were mixed with 1% formaldehyde for 10 min and 0.125 M glycine solution was added to end this reaction. Then, cells were rinsed using washing buffer and re-suspended in sonication buffer. Followed by sonication, the samples were mixed with immunoprecipitation buffer and incubated with relevant antibodies. For ChIP, the immune-precipitates were rinsed using washing buffer. Followed by reverse-crosslinking, the DNA samples were isolated and analyzed by real-time quantitative PCR. The primer sequences for p21 promoter were: 5′-GTGCGTCTGATTGCGTTTCTG-3′ (forward) and 5′-CTGAAAACGAGCAGCGGAAG-3′ (reverse). The primer sequences for puma promoter were: 5′-GCGGAACTGTGCGCTTGTGT-3′ (forward) and 5′-CGCTCAGGGCTCACAAAGT-3′ (reverse).
RT-PCR
Total RNAs were isolated using TRIzolTM Reagent (Invitrogen, Carlsbad, CA, USA). cDNA was reversely transcribed using GoScriptTM Reverse Transcriptase System (Promega, Fitchburg, WI, USA) in line with the supplier’s instruction. The expression levels of p21 and puma were measured using SYBR Green PCR Kit (Applied Biosystems, Foster City, CA, USA). The β-actin expression was utilized as internal control. The PCR primer sequences for p21 were: 5′-AAGCCTTGATTCTGATGTGGGC-3′ (forward) and 5′-TGACGAAGTCAAAGTTCCACCG-3′ (reverse). The PCR primer sequences for puma were: 5′-AGACAAGAAGAGCAGCATCGACAC-3′ (forward) and 5′-TAGGCACCTAGTTGGGCTCCATTT-3′ (reverse). The PCR primer sequences for β-actin were: 5′-GTGGGCCGCTCTAGGCACCAA-3′ (forward) and 5′-CTCTTTGATGTCACGCACGATTTC-3′ (reverse). Data were analyzed using 2-△△Ct method [Citation20].
Preparation of recombinant protein from bacteria
Flag-tagged WT, K-R mutated or K-Q mutated JHDM1A, 300 and core histones were all expanded by PCR, cloned into His-tag bacterial expression vector Pet30a (Novagen, Madison, WI, USA). The constructs were confirmed by DNA sequencing. Then, the plasmids were transfected into bacteria. To induce recombinant proteins expression, the bacteria was mixed with 0.1 mM IPTG at 37 °C for 4 h. Ni-NTA affinity column (Qiagen, Valencia, CA, USA) was utilized for isolated His-tagged recombinant proteins. Subsequently, the elute was dialyzed in PBS overnight at 4 °C (dialysis kits). The homogeneity and concentration of these proteins were tested on SDS-PAGE by Coomassie blue staining using bovine serum albumin (BSA) as a standard control.
In vitro acetylation assay
Flag-p300 and Flag-JHDM1A proteins were precipitated from transfected HEK293 cells with anti-Flag antibody. The acetylation reaction was carried out as earlier reported [Citation19]. After reaction, the reaction mixture was electrophoresed on SDS-PAGE gels for immune-blotting analysis.
Glutathione S-transferase (GST) pull down assay
GST pull-down assay was done as earlier described [Citation21]. Briefly, GST-tagged JHDM1A was expressed in Escherichia coli was mixed with GS-4B beads (GE healthcare, Beijing, China) and spun down at 4 °C. Followed by rinsing, the beads were reacted with in vitro-transcribed/translated HA-tagged p300. Finally, the p300 protein level was tested using Western blotting.
Mononucleosoma preparation
Mononucleosoma preparation was done in line with the previously described [Citation19]. The nuclear pellet isolated from about 1 × 108 cells were mixed with lysis buffer to obtain oligonucleosomes. Then, the oligonucleosomes were digested with micrococcal nuclease (10 units/ml) for 10 min at 37 °C. After inactivation with 5 mM EDTA, the nuclease was centrifuged at 14000 rpm for 5 min. The supernatant was subjected to 10–40% glycerol gradient sedimentation. The mononucleosomes were derived from these supernatants through testing aliquots of fractions (digested with Proteinase K) on a 1% DNA agarose gel.
Demethylase assay and nucleosome binding assay in vitro
Bulk histones or nucleosomes were mixed with purified recombinant His-JHDM1A (WT) or His-JHDM1A mutants (K-Q, K-R) in the histone demethylation (HDM) assay buffer for 2 h at 37 °C. For a typical reaction, the volume of the reaction is 100 μl, in which 2 μg bulk histones or 5 μg nucleosomes were used as substrates. The demethylase activity of JHDM1A was determined by SDS-PAGE and Western blotting detection with H3K36me2, H3K36me3 or JHDM1A antibodies.
Detection of cell viability
After indicated transfections, viability of HOS cells was detected using cell counting kit-8 (CCK-8) assay (Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s instruction. The OD-values of each group was detected with the help of Microplate reader (Bio-Tek Instruments, Winooski, VT, USA).
Colony formation assay
After indicated transfections, colony formation assay was conducted to assess the long-term proliferation activity of cells. Briefly, transfected HOS cells were cultivated into 6-well plate (500 cells/well) and grown at 37 °C with 5% for 2 weeks. Culture medium was changed every three days. The number of colonies in each group was counted.
5-bromo-2′-deoxyuridine (BrdU) incorporation assay
After different transfection, HOS cells were cultivated into 6-well plate (1 × 105 cells/well). BrdU solution was mixed into the culture medium for 24 h. Then, the percentage of BrdU positive cells was counted.
Wound closure assay
After different transfection, HOS cells were cultivated into a 6-well plate (4 × 105 cells/well) for 12 h. Then, 200 μl pipette tips were crossed to induce wounds. Culture medium was changed by serum-free MEM medium for 24 h. Wound closure was expressed as the percentage of original wound width.
Two chamber transwell assay
Cell invasion was assessed by two-chamber transwell assay (Corning Corporation, New York, NY, USA). Briefly, after different transfection, 1 × 103 HOS cells in 200 μl serum-free MEM medium were added into the upper chamber. Complete MEM medium (600 μl) was added into the lower chamber. After incubation for 48 h, invaded cells in the lower chamber were counted under a microscope. Data were presented as the percentage of original cells.
Mouse tumor models
Twenty-eight nude mice (8–9 weeks, 20.1–23.9 g body weight) were supplied by Beijing Huafukang Bioscience Co., Ltd., (Beijing, China). All experimental procedures were performed in accordance with the National Institute of Health’s Guide for the Care and Use of Laboratory Animals and approved by our local institution. Briefly, mice were randomly divided into four groups: Vector, WT, K-R and K-Q. Relevant transfected HOS cells (1 × 107) were injected into the hind limbs of mice. The long (a) and short (b) diameters of the tumors were tested every 5 days. Tumor volume was measured using formula ab2/2. After 35 days, the mice were intraperitoneally injected by 2% pentobarbital sodium (40 mg/kg body weight, Sigma-Aldrich) and sacrificed.
Statistical analysis
Data were presented as the mean ± standard deviation (SD) for three independent experiments. Statistical analyses were conducted using Graphpad 6.0 software (Graphpad, La Jolla, CA, USA). p Values were calculated by ANOVA. Statistical significance was set at p < .05.
Results
JHDM1A was acetylated by p300 in osteosarcoma cells
Firstly, whether JHDMI1 can be acetylated in osteosarcoma MG-63 and HOS cells were tested. Plasmid expressing Flag-tagged JHDM1A was transfected into MG-63 orHOS cells, respectively. Flag-JHDM1A proteins were immunoprecipitated from cells with anti-Flag antibody. Subsequently, immunoblotting with pan-acetyl-lysine antibody was carried out to test the acetylation of JHDM1A. Results showed that JHDM1A could be acetylated in MG-63 and HOS cells (). Moreover, histone deacetylase (HDAC) inhibitor TSA and/or sirtuin 1 (SIRT1) inhibitor NAM treatment promoted the acetylation of JHDM1A in MG-63 and HOS cells (). In order to identify the lysine acetyltransferase enzyme(s) related to JHDM1A acetylation in cells, a number of enzyme(s) from multiple lysine acetyltransferase families, including KAT2B, KAT8, Gcn5/PCAF, p300 and MYST were investigated. These acetyltransferases were co-expressed with Flag-JHDM1A, respectively and their abilities to stimulate JHDM1A acetylation were analyzed. Results displayed that only p300 strongly promoted JHDM1A acetylation (). Besides, sh-p300 was transfected into MG-63 and HOS cells to knock down p300 expression. displayed that the endogenous JHDMIA protein level was not changed after sh-p300 transfection, while the acetylation level of JHDM1A was reduced after sh-p300 transfection. Altogether, these results suggested that p300 could promote the acetylation of JHDM1A in osteosarcoma cells.
Figure 1. JHDM1A was acetylated by p300 in osteosarcoma cells. (A) MG-63 and HOS cells were transfected with plasmids expressing Flag-tagged JHDM1A, respectively. Immunoprecipitation (IP) assay with anti-Flag antibody and immunoblotting assay with pan-acetyl-lysine (Ac-K) antibody showed that JHDM1A could be acetylated in osteosarcoma MG-63 and HOS cells. TSA and NAM further promoted the acetylation of JHDM1A. (B) p300 and Flag-JHDM1A were co-transfected into HEK293 cells. IP assay with anti-Flag antibody and immunoblotting assay with Ac-K antibody showed that p300 induced JHDM1A acetylation. (C) sh-p300 was transfected into MG-63 and HOS cells. IP assay with anti-Flag antibody and immunoblotting assay with Ac-K antibody showed that knockdown of p300 reduced the acetylation of JHDM1A. (D) IP assay was carried out in HOS cell lysates using antibody against p300. Western blotting using JHDM1A antibody showed that JHDM1A also existed in the complex. (E) HA-tagged JHDM1A and Myc-tagged p300 were co-transfected into MG-63 cells. IP assay was carried out in MG-63 cell lysates using antibody against HA. Western blotting using anti-Myc antibody and anti-HA showed that p300 also existed in the complex. (F) Glutathione S-transferase (GST) pull down assay with GST-tagged JHDM1A and p300 showed that p300 directly interacted with JHDM1A.
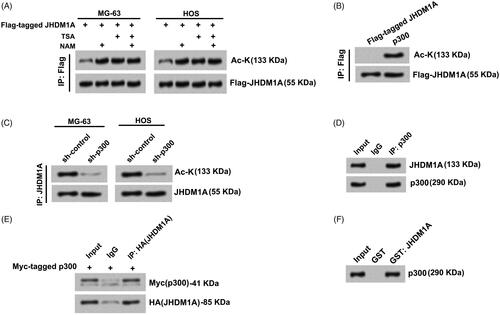
JHDM1A interacted with p300 in osteosarcoma cells
Next, whether JHDM1A directly interacted with p300 in osteosarcoma cells was probed. Endogenous p300 protein in HOS cells was immunoprecipitated using antibody against p300. Data in showed that JHDM1A was co-precipitated in the complex. In a similar assay, when plasmid expressing HA-tagged JHDM1A and Myc-tagged p300 was transfected into MG-63 cells. HA-tagged JHDM1A was immunoprecipitated using antibody against HA. p300 was also co-precipitated in the complex (). In addition, GST-tagged JHDM1A was expressed in Escherichia coli. In vitro-transcribed/translated HA-tagged p300 was purified and incubated with GST-JHDM1A. Result of GST pull-down assay in presented that p300 directly interacted with JHDM1A. These findings indicated that p300 could directly interact with JHDM1A and acetylate JHDM1A in osteosarcoma cells.
p300 directly acetylated JHDM1A at lysine residues 409 (K409)
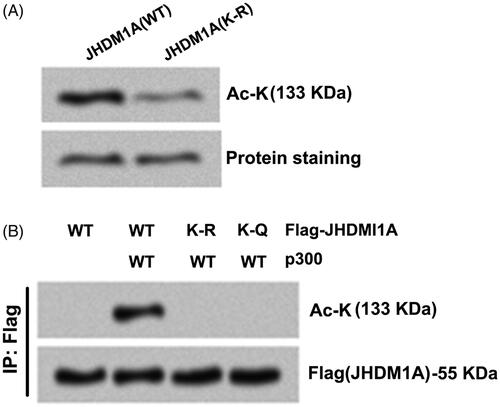
Proteomics analysis using Uniprot (https://www.uniprot.org) predicted that JHDM1A can be acetylated at K409 site. In order to analyze whether p300 acetylates JHDM1A at K409 site, recombinant full-length wild-type (WT) JHDM1A protein was purified from bacteria. Moreover, the K409 site of JHDM1A was replaced using arginine (R) to form acetylation-resistant (K-R mutated) JHDM1A. To prepare p300, Flag-p300 was expressed in HEK293 cells and p300 protein was immunoprecipitated using anti-Flag antibodies. showed that WT JHDM1A was acetylated by immunoprecipitates containing p300 in vitro. By contrast with WT JHDM1A, the acetylation level of JHDM1A was lowered in K-R mutated JHDM1A group when incubating with immunoprecipitates containing p300 (). Moreover, the K409 site of JHDM1A was replaced using glutamine (Q) to form acetylation-mimicking (K-Q mutated) JHDM1A. displayed that followed by p300 and Flag-tagged WT (or K-R, K-Q mutant) JHDM1A were co-transfected into HEK293 cells, the acetylation levels of JHDM1A were both abolished in K-R mutated JHDM1A group and K-Q mutated JHDM1A group (). These results indicated that p300 directly acetylated JHDM1A at lysine residues 409.
Figure 2. p300 directly acetylated JHDM1A at lysine residues 409 (K409). (A) K409 site of JHDM1A was replaced using arginine (R) to form acetylation-resistant (K-R mutated) JHDM1A. p300 immuno-precipitates (from Flag-p300 expressed in HEK293 cells) and recombinant full-length wild-type (WT) JHDM1A protein or K-R mutated JHDM1A protein (from bacteria) were co-incubated. Immunoblotting assay with pan-acetyl-lysine (Ac-K) antibody showed that the K-R mutated JHDM1A was resistant to p300-mediated acetylation. (B) K409 site of JHDM1A was replaced using glutamine (Q) to form acetylation-mimicking (K-Q mutated) JHDM1A. p300 and Flag-tagged WT (or K-R, K-Q mutated) JHDM1A were co-transfected into HEK293 cells. Immunoprecipitation (IP) assay with anti-Flag antibody and immunoblotting assay with Ac-K antibody showed that both K-R JHDM1A and K-Q JHDM1A were resistant to p300-mediated acetylation.
Acetylation of JHDM1A disrupted its association with nucleosomes and demethylation of H3K36me2
Subsequently, whether K-Q mutated JHDM1A can bind nucleosomes in vitro was explored. Mononucleosomes were reconstituted using recombinant core histones and biotin-tagged DNA fragments. Then, the nucleosome particles were mixed with recombinant WT or K-Q mutated JHDM1A proteins. The associated proteins were tested by immunoblotting. Results showed that the binding of WT JHDM1A to nucleosomes became readily detected (). By contrast, the binding of K-Q mutated JHDM1A to nucleosomes was reduced (), which suggested that acetylation of JHDM1A reduced its ability to binding nucleosomes.
Figure 3. Acetylation of JHDM1A disrupted its binding to nucleosomes and demethylation of H3K36me2. (A) Recombinant core histones and biotin-labeled DNA (Bio-nucl) were utilized for reconstituting nucleosome. WT or K-Q mutated JHDM1A was mixed with reconstituted nucleosomes. Immunoblotting assay with anti-JHDM1A antibody showed that the binding of K-Q mutated JHDM1A to nucleosomes was reduced in vitro. (B) WT or K-Q, K-R mutated JHDM1A was incubated with bulk histone. Immunoblotting assay with anti-H3K36me2 antibody, anti-H3K36me3 antibody or anti-JHDM1A antibody showed that the acetylation status of JHDM1A had no influences on its ability to demethylate H3K36 in bulk histones in vitro. H3K36me3 level was used as a loading control. (C) WT or K-Q, K-R mutated JHDM1A was incubated with mononucleosomes isolated from MG-63 cells. Immunoblotting assay with anti-H3K36me2 antibody, anti-H3K36me3 antibody or anti-JHDM1A antibody showed that the K-Q mutated JHDM1A could not demethylate H3K36 in mononucleosomes. H3K36me3 level was used as a loading control.
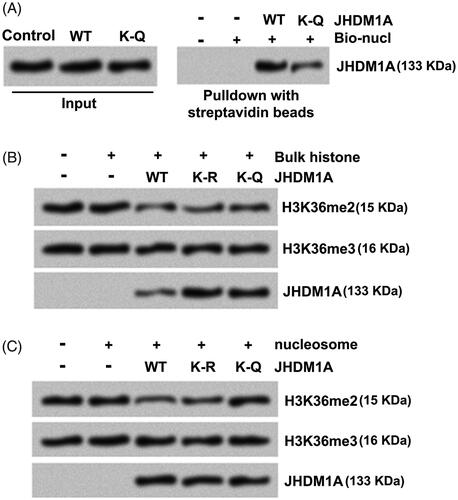
JHDM1A alone can demethylate H3K36me2 in peptides or bulk histones. Whether acetylation of JHDM1A reduces its ability to demethylate H3K36me2 was probed. Data in displayed that when mixed with bulk histones, recombinant WT, K-R or K-Q mutated JHDM1A proteins all decreased H3K36me2 levels, which implied that the acetylation status of K409 in JHDM1A had no influence on its enzymatic activity on free histones. However, when mixed with mononucleosomes isolated from cells, WT and K-R mutated JHDM1A demethylated H3K36me2 in nucleosomes, while K-Q mutated JHDM1A failed to demethylated nucleosomal H3K36me2 (). These findings indicated that the demethylation of H3K36me2 was reduced after acetylation of JHDM1A in cells.
Acetylation of JHDM1A impaired its ability to repress p21 and puma transcription
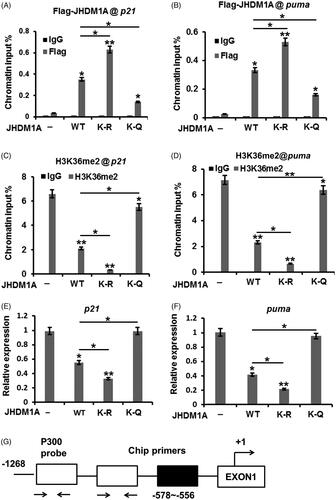
Chromatin immunoprecipitation (ChIP) analysis was carried out to probe whether acetylation of JHDM1A influences its association on genomic targets. showed that WT JHDM1A could bind to the p21 and puma promoters (p < .05 or p < .01), while K-Q mutated JHDM1A notably lowered its association with the p21 and puma promoters (p < .05), which suggested that acetylation of JHDM1A attenuated its recruitment to chromatin in cells. K-R mutated JHDM1A exhibited stronger chromatin binding than WT JHDM1A (, p < .05). In addition, the H3K36me2 levels at the p21 and puma promoters were high in JHDM1A-null cells reconstituted with empty vector (). Introduction of WT or K-R mutated JHDM1A potently reduced H3K36me2 levels at these loci (, p < .05 or p < .01). By contrast, the K-Q JHDM1A could not reduce H3K36me2 levels at these loci (), which was consistent with its reduced association with chromatin. Besides, the p21 and puma mRNA levels were also detected after WT, K-R or K-Q mutated JHDM1A transfection. displayed that WT or K-R mutated JHDM1A transfection lowered the p21 and puma mRNA levels in MG-63 cells (p < .05 or p < .01), while K-Q JHDM1A had no significant influences on p21 and puma mRNA levels in MG-63 cells. These results suggested that acetylation of JHDM1A de-repressed the transcription of p21 and puma. The possible schematic presentation of chip regions and PCR primer binding sites was shown in .
Figure 4. Acetylation of JHDM1A impaired its ability to repress p21 and puma transcription. (A and B) Bindings of Flag-tagged WT or K-R, K-Q mutated JHDM1A proteins to the p21 and puma promoters were evaluated by ChIP analysis with anti-Flag antibodies. The bindings between K-Q mutated JHDM1A and p21 or puma promoters were notably reduced. (C and D) Followed by Flag-tagged WT or K-R, K-Q mutated JHDM1A transfection, the bindings of H3K36m2 to the p21 and puma promoters were measured by ChIP analysis. K-Q mutated JHDM1A failed to reduce the binding H3K36m2 to the p21 and puma promoters (E and F) Followed by Flag-tagged WT or K-R, K-Q mutated JHDM1A transfection, the p21 and puma mRNA levels in MG-63 cells were tested by real-time RT-PCR. (G) The possible schematic presentation of chip regions and PCR primer binding sites. *p ˂ .05; **p ˂ .01.
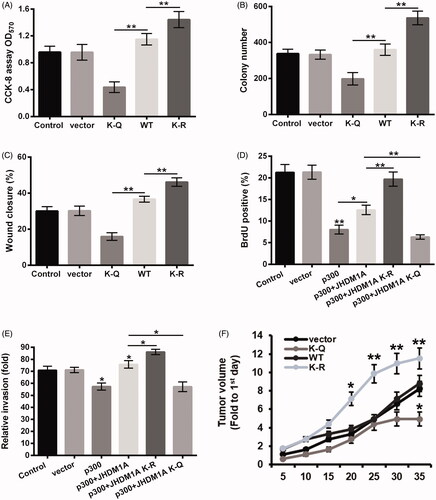
Acetylation of JHDM1A inhibited osteosarcoma carcinogenesis
Finally, the influence of JHDM1A K409 acetylation on osteosarcoma carcinogenesis was explored. displayed that compared to WT JHDM1A group, the viability and colony number of HOS cells were both decreased in K-Q mutated JHDM1A group (p < .01) and increased in K-R mutated JHDM1A group (p < .01). The results of wound healing assay in showed that K-R mutated JHDM1A promoted the migration of HOS cells (p < .01), while K-Q mutated JHDM1A had an opposite effect (p < .01). Furthermore, presented that p300 transfection inhibited the proliferation of HOS cells (p < .01). Relative to 300 + WT JHDM1A group, the rate of BrdU positive cells was enhanced in shp300 + JHDM1A K-R mutated group (p < .01) and reduced in shp300 + JHDM1A K-Q mutated group (p < .01). The results of the transwell assay in displayed that p300 transfection lowered the invasion of HOS cells (p < .05). WT or K-R mutated JHDM1A rescued the invasion capability of HOS cells (p < .05), but K-Q mutated JHDM1A had no significant effect on 300-induced HOS cell invasion inhibition. Besides, presented that K-R mutated JHDM1A notably promoted the tumor growth in mice (p < .05 or p < .01), while K-Q mutated JHDM1A inhibited the tumor formation in mice (p < .05). Altogether, these above results suggested that acetylation of JHDM1A could inhibit the proliferation, migration and invasion of osteosarcoma cells, as well as suppressed the tumor growth of osteosarcoma.
Figure 5. Acetylation of JHDM1A inhibited osteosarcoma carcinogenesis. Plasmid expressed WT, K-Q or K-R mutated JHDM1A was transfected into HOS cells, respectively. Empty plasmid was used as a negative control. (A) Cell viability, (B) colony formation and (C) cell invasion were tested using CCK-8 assay, colony formation assay and wound closure assay, respectively. Plasmid expressed p300 and plasmid expressed WT, K-Q or K-R mutated JHDM1A were co-transfected into HOS cells, respectively. Empty plasmid was used as a negative control. (D) Cell proliferation and (E) cell invasion were tested using 5-bromo-2′-deoxyuridine (BrdU) incorporation assay and two chamber transwell assay, respectively. (F) HOS cells were transfected with HA-tagged WT, K-Q or K-R mutated JHDM1A-expressing plasmids and injected into the hind limbs of mice to form mouse tumor models. The tumor volumes were measured every 5 days after injection. *p ˂ .05; **p ˂ .01.
Discussion
JHDM1A has been found to participate in cancer development via demethylate H3K36me2 [Citation10,Citation15]. In this research, we discovered that p300 directly acetylated JHDM1A at K409 residue in osteosarcoma cells. Moreover, acetylation of JHDMIA disrupted its binding with nucleosomes and thereby impaired its ability to demethylate H3K36me2 and derepressed p21 and puma transcription. Furthermore, acetylation of JHDM1A inhibited the carcinogenesis of osteosarcoma.
Dysregulation of histone methyltransferases and demethylases has been considered as an important epigenetic mechanism related to cancer onset and progression [Citation22]. As a histone demethylase, JHDM1A has been demonstrated to be up-regulated in many cancer cells, such as non-small cell lung cancer [Citation10], ovarian cancer [Citation16], breast cancer [Citation23] and gastric cancer [Citation24]. p300 is an acetyltransferase in cells, which can acetylate many proteins in cells and take part in the modulation of many cellular biological processes, including cell cycle transition [Citation25], DNA synthesis [Citation26], cell autophagy [Citation27], cell proliferation [Citation28] and cell apoptosis [Citation29]. The tumor suppressive role of p300 has been widely studied [Citation30]. p300 gene mutations, especially mutations at the sequence encoding the acetyltransferase, have been discovered to exist in many human cancers [Citation31]. In terms of osteosarcoma, Sui et al. reported that p300 exhibit tumor suppressive activity via acetylating p53 and then up-regulating p21 and Bax expression [Citation32]. Herein, we found that p300 could interact with JHDM1A and directly acetylate JHDM1A at K409 residue in osteosarcoma MG-63 and HOS cells, which implied that p300 exert tumor suppressive roles in osteosarcoma cells might be via acetylating JHDM1A.
H3K36me2 is an important histone modification pathway in cells [Citation33]. It exerts critical function in maintaining genomic stability and participates in the regulation of DNA repair [Citation9,Citation33]. Aberrant demethylation of H3K36me2 has been demonstrated to contribute to cancer development [Citation34,Citation35]. JHDM1A can demethylate H3K36me2 and play a tumor-promoting role in tumor cells [Citation10,Citation35]. Herein, we discovered that acetylation of JHDM1A (that could cause by p300) disrupted its binding to nucleosomes and thereby impaired its ability to demethylate H3K36me2. More importantly, the transcription of p21 and puma were both increased in osteosarcoma cells after mimicking of JHDM1A acetylation, which induced the mRNA expression levels of p21 and puma were enhanced in osteosarcoma cells. p21 is a cyclin-dependent kinase (CDKs) inhibitor that can cause cell-cycle arrest by interacting with different stimuli such as p53, CDKs, proliferating cell nuclear antigen (PCNA) and estrogen receptor-α (ER-α) [Citation36]. Puma is a member of the “BH3-only” branch of the Bcl-2 family localized in the mitochondria [Citation37]. It can trigger mitochondrial dysfunction-mediated cell apoptosis through antagonizing the functions of Bcl-xl and myeloid cell leukemia-1 (Mcl-1) [Citation38]. Both p21 and puma are lowly expressed and present tumor suppressive activities in cancer cells, including osteosarcoma cells [Citation39,Citation40]. Herein, we found that acetylation of JHDM1A up-regulated the mRNA expression levels of p21 and puma in osteosarcoma cells, which implied that p300-dependent acetylation of JHDM1A could inhibit osteosarcoma carcinogenesis. Moreover, further experiments were done to evaluate the acetylation of JHDM1A on osteosarcoma cell proliferation and invasion, as well as tumor growth. We found that acetylation of JHDM1A suppressed the proliferation and invasion of osteosarcoma HOS cells, as well as inhibited the tumor growth in vivo.
In conclusion, this research revealed the regulatory activity of p300 on JHDM1A acetylation in osteosarcoma cells. p300 could acetylate JHDM1A at K409 residue, which reduced the demethylation of H3K36me2, enhanced the transcription of p21 and puma and thereby inhibited the growth and metastasis of osteosarcoma. We propose that JHDM1A exerted an oncogenic role in osteosarcoma cells by demethylating H3K36me2, while p300 exerted tumor suppressive role in osteosarcoma cells at least partially via acetylating JHDM1A.
Acknowledgements
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Disclosure statement
The authors declare no conflict of interest.
References
- Harrison DJ, Geller DS, Gill JD, et al. Current and future therapeutic approaches for osteosarcoma. Expert Rev Anticancer Ther. 2018;18:39–50.
- Chrun ES, Modolo F, Daniel FI. Histone modifications: a review about the presence of this epigenetic phenomenon in carcinogenesis. Pathol Res Pract. 2017;213:1329–1339.
- Vaidya H, Rumph C, Katula KS. Inactivation of the WNT5A alternative promoter B is associated with DNA methylation and histone modification in osteosarcoma cell lines U2OS and SaOS-2. PLOS One. 2016;11:e0151392.
- La Noce M, Paino F, Mele L, et al. HDAC2 depletion promotes osteosarcoma's stemness both in vitro and in vivo: a study on a putative new target for CSCs directed therapy. J Exp Clin Cancer Res. 2018;3;37:296.
- Kelso TWR, Porter DK, Amaral ML, et al. Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. Elife. 2017;6:pii:e30506.
- Soci UPR, Melo SFS, Gomes JLP, et al. Exercise training and epigenetic regulation: multilevel modification and regulation of gene expression. Adv Exp Med Biol. 2017;1000:281–322.
- Brennecke P, Arlt MJ, Campanile C, et al. CXCR4 antibody treatment suppresses metastatic spread to the lung of intratibial human osteosarcoma xenografts in mice. Clin Exp Metastasis. 2014;31:339–349.
- Peng YB, Fan B, Han XL, et al. Molecular characterization of the porcine JHDM1A gene associated with average daily gain: evaluation its role in skeletal muscle development and growth. Mol Biol Rep. 2011;38:4697–4704.
- Fnu S, Williamson EA, De Haro LP, et al. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc Natl Acad Sci USA. 2011;108:540–545.
- Dhar SS, Alam H, Li N, et al. Transcriptional repression of histone deacetylase 3 by the histone demethylase KDM2A is coupled to tumorigenicity of lung cancer cells. J Biol Chem. 2014;289:7483–7496.
- Hsia DA, Tepper CG, Pochampalli MR, et al. KDM8, a H3K36me2 histone demethylase that acts in the cyclin A1 coding region to regulate cancer cell proliferation. Proc Natl Acad Sci USA. 2010;107:9671–9676.
- Sankaran SM, Gozani O. Characterization of H3.3K36M as a tool to study H3K36 methylation in cancer cells. Epigenetics. 2017;12:917–922.
- Tanaka Y, Yano H, Ogasawara S, et al. Mild glucose starvation induces KDM2A-mediated H3K36me2 demethylation through AMPK to reduce rRNA transcription and cell proliferation. Mol Cell Biol. 2015;35:4170–4184.
- Varier RA, Timmers HT. Histone lysine methylation and demethylation pathways in cancer. Biochim Biophys Acta. 2011;1815:75–89.
- Wagner KW, Alam H, Dhar SS, et al. KDM2A promotes lung tumorigenesis by epigenetically enhancing ERK1/2 signaling. J Clin Invest. 2013;123:5231–5246.
- Lu DH, Yang J, Gao LK, et al. Lysine demethylase 2A promotes the progression of ovarian cancer by regulating the PI3K pathway and reversing epithelial mesenchymal transition. Oncol Rep. 2019;41:917–927.
- Chen J, Ghazawi FM, Li Q. Interplay of bromodomain and histone acetylation in the regulation of p300-dependent genes. Epigenetics. 2010;5:509–515.
- Holmqvist PH, Mannervik M. Genomic occupancy of the transcriptional co-activators p300 and CBP. Transcription. 2013;4:18–23.
- Luo H, Shenoy AK, Li X, et al. MOF acetylates the histone demethylase LSD1 to suppress epithelial-to-mesenchymal transition. Cell Rep. 2016;15:2665–2678.
- Ish-Shalom S, Lichter A. Analysis of fungal gene expression by real time quantitative PCR. Methods Mol Biol. 2010;638:103–114.
- Machida K, Liu B. Binding assays using recombinant SH2 domains: far-western, pull-down, and fluorescence polarization. Methods Mol Biol (Clifton, NJ). 2017;1555:307–330.
- Morera L, Lubbert M, Jung M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin Epigenet. 2016;8:57.
- Rizwani W, Schaal C, Kunigal S, et al. Mammalian lysine histone demethylase KDM2A regulates E2F1-mediated gene transcription in breast cancer cells. PLOS One. 2014;9:e100888.
- Huang Y, Liu Y, Yu L, et al. Histone demethylase KDM2A promotes tumor cell growth and migration in gastric cancer. Tumor Biol. 2015;36:271–278.
- Rajabi HN, Baluchamy S, Kolli S, et al. Effects of depletion of CREB-binding protein on c-Myc regulation and cell cycle G1-S transition. J Biol Chem. 2005;280:361.
- Sankar N, Kadeppagari RK, Thimmapaya B. c-Myc-induced aberrant DNA synthesis and activation of DNA damage response in p300 knockdown cells. J Biol Chem. 2009;284:15193–15205.
- Wan W, You Z, Xu Y, et al. mTORC1 Phosphorylates acetyltransferase p300 to regulate autophagy and lipogenesis. Mol Cell. 2017;68:323–335.e6.
- Sandberg ML, Sutton SE, Pletcher MT, et al. c-Myb and p300 regulate hematopoietic stem cell proliferation and differentiation. Dev Cell. 2005;8:153–166.
- Gao XN, Lin J, Ning QY, et al. A histone acetyltransferase p300 inhibitor C646 induces cell cycle arrest and apoptosis selectively in AML1-ETO-positive AML cells. PLOS One. 2013;8:e55481.
- Suganuma T, Ikeda MA. Tumor growth suppression by the coactivator p300. J Oral Biosci. 2008;50:115–124.
- Cho YA, Hong JS, Choe EJ, et al. The role of p300 in the tumor progression of oral squamous cell carcinoma. J Oral Pathol Med. 2015;44:185–192.
- Sui Y, Han Y, Zhao X, et al. Long non-coding RNA GClnc1 promotes tumorigenesis in osteosarcoma by inhibiting p53 signaling. Biochem Biophys Res Comm. 2018;507:36–42.
- Amendola PG, Zaghet N, Ramalho JJ, et al. JMJD-5/KDM8 regulates H3K36me2 and is required for late steps of homologous recombination and genome integrity. PLOS Genet. 2017;13:e1006632.
- Zheng Q, Fan H, Meng Z, et al. Histone demethylase KDM2B promotes triple negative breast cancer proliferation by suppressing p15INK4B, p16INK4A, and p57KIP2 transcription. Acta Biochim Biophys Sinica. 2018;50:897–904.
- He J, Nguyen AT, Zhang Y. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood. 2011;117:3869–3880.
- Parveen A, Akash MS, Rehman K, et al. Dual role of p21 in the progression of cancer and its treatment. Crit Rev Eukaryot Gene Expr. 2016;26:49–62.
- Yang J, Zhao X, Tang M, et al. The role of ROS and subsequent DNA-damage response in PUMA-induced apoptosis of ovarian cancer cells. Oncotarget. 2017;8:23492–23506.
- Woo TG, Yoon MH, Hong SD, et al. Anti-cancer effect of novel PAK1 inhibitor via induction of PUMA-mediated cell death and p21-mediated cell cycle arrest. Oncotarget. 2017;8:23690–23701.
- Ruan J, Zheng L, Hu N, et al. Long noncoding RNA SNHG6 promotes osteosarcoma cell proliferation through regulating p21 and KLF2. Arch Biochem Biophys. 2018;646:128–136.
- Jiang K, Zhang C, Yu B, et al. Autophagic degradation of FOXO3a represses the expression of PUMA to block cell apoptosis in cisplatin-resistant osteosarcoma cells. Am J Cancer Res. 2017;7:1407–1422.