
Abstract
Researchers have been exploring the genetic mechanisms underlying the control of liver regeneration (LR). However, an integrated analysis of circRNAs expression of rat regenerating livers during the proliferation phase has not been performed yet. For this purpose, circRNAs expression profile was globally analyzed by high-throughput sequencing. It showed that 10,003 circRNAs were detected, and 164 circRNAs were differentially expressed. Subsequently, 27 circRNAs were predicted to bind to 58 candidate miRNAs and compete for miRNA-binding sites with 2195 mRNAs. By applying GO and KEGG analysis, it was predicted that these circRNAs significantly participated in tissue regeneration, regulation of cell proliferation and Ras, p53, Wnt, Jak-STAT, MAPK signalling pathways. Based on the number of the corresponding miRNAs and their role enriched and reported in cell proliferation of LR or hepatocellular carcinoma, four kinds of circRNAs (circ_03848, circ_08236, circ_13398 and circ_15013) were considered as the key circRNAs. The predicted competing endogenous RNA networks and bioinformatics analysis revealed the potential role of these circRNAs in LR, which would provide useful information for understanding the mechanism of LR.
Introduction
The liver is a vital organ for detoxification, protein synthesis, digestion and other functions, and its regenerative capacity is powerful in animals. 2/3 partial hepatectomy (PH) in rats is an ideal model for liver regeneration (LR) [Citation1]. The process of LR is generally divided into three phases: priming (0–6 h after PH), proliferation (12–72 h after PH) and termination (72–168 h after PH) [Citation2]. Hepatic cells are activated from G0 phase to G1 phase in the priming phase [Citation3]. Especially, activated hepatic cells enter S phase and perform two cell cycles in the proliferation phase. A number of studies were conducted to reveal the genetic mechanism regulating the second phase of LR, for instance, hepatocyte growth factor (HGF), mitosis-promoting factors such as epidermal growth factor (EGF) and transforming growth factor alpha play an important role in this phase [Citation4]. However, the mechanism is not yet completely clear.
circRNAs were discovered in 1976, and are predominantly generated by back-splicing reactions, in which the 5′ and 3′ ends of linear RNAs are directly ligated [Citation5], and are widely found in many kinds of tissues and cells, and own the characteristics of stable structure, conservative sequence and specificity of expression pattern. According to composition and different sources, circRNAs can be divided into the following types: exonic circRNA, intronic circRNA (circular intronic RNAs), intergenic circRNA, antisense circRNA and sense-overlapping circRNA [Citation6].
It has been verified that circRNAs work through various ways including acting as miRNAs sponge, regulating transcription by interacting with small nuclear ribonucleoproteins (snRNP), recruiting RNA polymeraseII, interacting with proteins, and coding protein [Citation7]. We had shown that 159 circRNAs were differentially expressed (DE) in the priming phase after PH [Citation8]. In the present study, we aimed primarily to investigate the potential mechanism of circRNAs regulating the proliferation phase after 2/3 PH in rats.
Materials and methods
Preparation of rat LR model
Male Sprague–Dawley rats (age 8–10 weeks, body weight 200–220 g) were obtained from Laboratory Animal Center of Zhengzhou University (Zhengzhou, China). Animals were housed in standard cages for one week at least before the experiments. The animals were kept at a constant temperature of 22 ± 1 °C, relative humidity of 55–65% and a 12 h light–dark cycle. 2/3 PH was performed on adult male rats (10 to 12 weeks old) as previously described by Higgins [Citation9] using ether anaesthesia. At 0, 12, 24, 30, 36 and 72 h after PH, the right liver lobes from six rats at each time point after PH were pooled to account for biological variation between 9:00 and 11:00 am, and stored at −80 °C after liquid nitrogen freezing. Excessive inhalation of CO2 was used as the euthanasia methods after the experiment. All experimental procedures in this study were approved by and performed according to the guidelines for the care and use of experimental animals that have been established by the Animal Care and Use Committee of the Ministry of Science of the People’s Republic of China.
High-throughput RNA sequencing
All harvested liver tissues were sent to OE Biotech (Shanghai, China) for the whole transcriptome sequencing and quantitative analysis. Briefly, total RNA was extracted from each sample at 0, 12, 24, 30, 36 and 72 h after PH by using TRIzol reagent (Invitrogen, USA) following the manufacturer’s protocol. RNA integrity was evaluated using the Agilent 2100 Bioanalyser (Agilent Technologies, Santa Clara, CA, USA). The samples with RNA Integrity Number ≥ 7 were subjected to the subsequent analysis. The libraries were constructed using TruSeq Stranded Total RNA with Ribo-Zero Gold (Illumina, San Diego, CA, USA) and Ribonuclease R according to the manufacturer’s instructions. Then these libraries were sequenced on the Illumina sequencing platform (HiSeqTM 2500 or other platform) and 150/125 bp paired-end reads were generated. The reads were mapped, quantified and annotated as described by Li et al. [Citation8]. Briefly, the clean reads were aligned to the reference genome by Bowtie2. And for unmapped reads, the junctions were picked out using back-splice algorithm. circRNAs were verified with a software developed by OE which were considered as the reference sequence for further analysis. Finally, the expression level of circRNAs was measured by RPM (mapped backsplicing junction reads per million mapped reads, RPM = number of circular reads/number of totalreads (units in million)).
Identification of DE circRNAs
To assess the changes of circRNA expression during the proliferation phase, DE circRNAs were defined by two statistical criteria (|log2Foldchange| ≥ 1 and p values ≤ .05). It was performed by the negative binomial distribution test based on the DESeq package in the same circRNA in each group (12, 24, 30, 36 and 72 h after PH) compared to normal liver tissue (0 h after PH), which was used to control filtration upon the statistics of alignment quality scores (the number of junction reads counts of circRNA in each sample was standardized). The hierarchical heatmap was performed to show the circRNA expression pattern among samples by Cluster 3.0.
Target miRNAs prediction of circRNAs
More and more researcher reported that circRNAs act as miRNA sponges by directly targeting miRNA to influence related miRNA activities. The candidate target miRNAs of circRNAs were predicted by miRanda [Citation10]. Interaction between miRNAs and circRNAs were defined with a maximum binding Max energy of –30 and a minimum Max score of 150. Free energy refers to the energy released when circRNA formed a stable structure with miRNA, the lower value of free energy indicated the stronger binding ability. Hypergeometric distribution assay was used to identify miRNAs that had a significant effect on circRNA during the proliferation phase of LR. While smaller p-values of enrichment indicated DE circRNAs was enriched in the corresponding miRNA.
Downstream mRNA prediction of miRNAs
As a small endogenous non-coding RNAs, miRNAs mediate post-transcriptional gene silencing by guiding Argonaute (AGO) proteins to RNA targets [Citation11]. For high-throughput experimental studies, it is a popular way that miRNA function was defined by its predicted downstream mRNA using TargetScan (http://www.targetscan.org) according to the threshold of total context++ score ≤ −0.6 [Citation12].
GO and KEGG enrichment analysis of the downstream mRNAs
To reveal the potential role of DE circRNAs, their downstream mRNAs of the candidate target miRNAs were submitted to the DAVID website (http://david.abcc.ncifcrf.gov) for the GO and KEGG enrichment analysis [Citation13]. The enriched GO terms were presented by enrichment score. KEGG enrichment analysis was used to determine the involvement of downstream mRNAs in different biological pathways. The –log10 (p-values) yielded an enrichment score indicating the significance of GO term and KEGG pathway correlations.
Construction of circRNA–miRNA–mRNA networks
In recent years, there is an increasing amount of evidence that circRNAs act as miRNA sponges [Citation14], and miRNAs cause post-transcriptional gene silencing by base pairing with mRNA for degradation and/or translational repression [Citation15]. The miRNA expression profiles in liver injury were reported [Citation16,Citation17]. It has been demonstrated that miRNAs played an important role in LR [Citation18]. One circRNA can sponge a number of target miRNA, and a target miRNA can be regulated by multiple circRNAs. Similarly, a miRNA can silence a number of target genes, and a target gene can be regulated by multiple miRNAs. Thus, to well elucidate the possible function of DE circRNAs during the proliferation phase, the network of circRNA–miRNA–mRNA is necessary. Complying with the above criterion, circRNA–miRNA–mRNA networks were displayed by Cytoscape 3.2.
Screening of key circRNAs
A combination of deep sequencing and bioinformatics analysis is a more effective way to identify key circRNAs and their potential function in the rat liver. The selection of key circRNAs among DE circRNAs was carried out according to the following principles. The number of nodes in the circRNA–miRNA network was not less than 3. Moreover, give priority to circRNAs that target the miRNAs associated with cell proliferation in rat LR or hepatocellular carcinoma (HCC) that were confirmed by experiments.
Validation of candidate key circRNA by qRT-PCR
To validate the data from high-throughput sequencing, the candidate key circRNAs were amplified with specific divergent primer in the PH groups and the control groups. Each pair divergent primers were designed according to the corresponding linear transcript sequence and synthesized in Sango Biotech (Shanghai, China). Specific primers designed for circRNAs are listed in . Total RNAs from regenerating liver and normal tissues were extracted using the TRIzol reagent (Invitrogen, USA) according to the manufacturer’s protocol. RNA quality was estimated by spectrophotometry and agarose gel electrophoresis. Then the total RNAs was incubated 1 h at 37 °C with 20 units of RNase R (Epicentre Biotechnologies). RNA was subsequently purified by phenol-chloroform extraction. 1 μg of RNA per sample was reverse transcribed into cDNA using a cDNA Reverse Transcription Kit (Takara, Tokyo, Japan). The PCR program was as follows: 95 °C for 1 min, followed by 40 cycles of 95 °C for 30 s, and 60 °C for 30 s and 72 °C for 45 s. Expression of circRNAs was normalized to GAPDH (internal standard control) and calculated using the 2−ΔΔCt method. All qRT-PCRs were performed in triplicate.
Table 1. Sequences of the out-facing primers used in this study.
Statistical analysis
Statistical analysis was performed with software SPSS 17.0. The data were presented as mean ± SEM. Group comparisons were evaluated by one-way ANOVA to determine statistical significance. Differences were statistically significant when p values ≤.05.
Results
Genomewide identification of circRNAs during the proliferation phase of rat LR
To investigate circRNAs at a genome-wide level, the total RNAs was isolated from six rat livers. After eliminating ribosome RNAs (rRNAs) and treating with RNase R, it was utilized to construct libraries for deep sequencing. The sequencing data reached 18.02 Gnt, 21.07 Gnt, 18.14 Gnt, 15.59 Gnt, 14.92 Gnt and 17.96 Gnt (Giga nucleotides, Gnt) raw bases in total from 0, 12, 24, 30, 36 and 72 h after PH. It covered 120.15 M, 140.49 M, 120.99 M, 103.98 M, 99.48 M, 119.73 M paired-end individual reads sized above 150 nt. After trimming adaptors and filtering low-quality reads, we obtained 88.76 M, 89.50 M, 89.65 M, 85.65 M, 87.35 M and 88.70 M clean reads in above samples in the same order (; Additional file S1).
Table 2. High-throughput RNA sequencing data of rat liver after PHa.
circRNAs expression pattern during the proliferation phase of rat LR
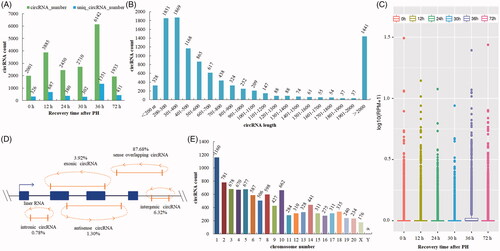
The whole transcriptome sequencing was used to detect circRNA expression profile using high-throughput sequencing and the Circbase database [Citation8]. A total of 10,003 unique circRNAs were identified in all samples (Additional file S2). The total number of detected circRNAs in each sample was as follows: 2001 (0 h), 3885 (12 h), 2450 (24 h), 2710 (30 h), 6142 (36 h) and 1933 (72 h). The unique number of circRNAs in the same order was 326, 687, 380, 302, 1351 and 431, respectively (). The size of the circRNAs ranged from 75 nt to 98,991 nt with an average size about 2600 nt. 14.41% of circRNAs had the predicted spliced length of more than 2000 nt. circRNAs with the length of less than 500 nt were 52.14%, and the length from 501 nt to 2000 nt were 33.45% (). Expression of circRNAs in all samples was measured based on RPM (mapped back-splicing junction reads per million mapped reads), which indicated that there was no abnormal expression in the six groups (). circRNAs were classified into five categories according to their relation with protein-coding genes: 1.30% circRNAs were antisense, 3.92% were exonic, 6.32% were intergenic, 0.78% were intronic, and 87.68% were sense overlapping (). They were widely distributed in all chromosomes ().
Figure 1. Expression pattern of circRNAs was detected by high-throughput RNA sequencing during the proliferation phase of rat LR. (A) circRNAs detected at each time point in the proliferation phase of LR. (B) The length distribution of circRNAs. (C) Box plots of RPM value of circRNAs. (D) The category of circRNAs. (E) CircRNA distribution on chromosome/scaffold.
Identification of DE circRNAs during the proliferation phase of rat LR
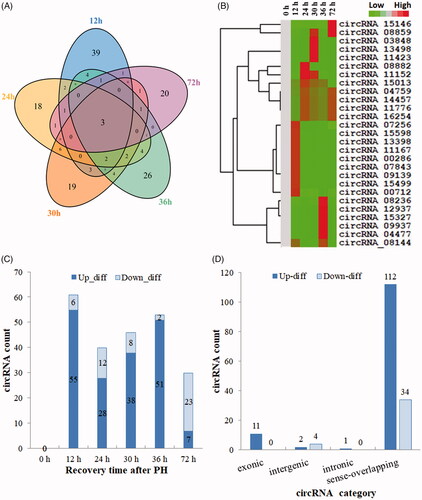
In this study, the following criteria: |log2Foldchange| ≥1 and p-values ≤.05 were used to identify the DE circRNAs, which performed by the negative binomial distribution test. As a result, 164 DE circRNAs were found between the normal (0 h) and regenerating livers (Additional file S3), which suggested that multiple circRNAs were involved in LR. Venn diagram displayed the DE circRNAs with statistical significance between regenerating and normal livers (). Hierarchical clustering exhibited the circRNA expression pattern in regenerating livers (). 126 out of 164 DE circRNAs were upregulated while 38 were downregulated. Thereinto, the number of upregulated circRNAs at 12, 24, 30, 36 and 72 h after PH was 55, 28, 38, 51 and 7. And the number of downregulated circRNAs in the same order was 6, 12, 8, 2 and 23, respectively (). According to different composition and sources, the upregulated circRNAs were divided into 1 intergenic, 112 sense overlapping, 1 intronic, and 11 exonic. On the other hand, the downregulated circRNAs were divided into 34 sense overlapping and 4 intronic ().
Figure 2. Identification of DE circRNAs in the proliferation phase of LR. (A) Venn analysis of DE circRNAs. (B) Hierarchical cluster analysis and heatmap. (C) The expression situation of DE circRNAs. (D) The distribution of DE circRNAs in different categories.
Bioinformatics analysis of competing endogenous RNA (ceRNA) networks during the proliferation phase of rat LR
circRNAs can affect gene expression at post-transcriptional level by acting as miRNAs sponge [Citation14]. According to the threshold of max energy ≤ −30 and max score ≥ 150, 35 out of 164 DE circRNAs were predicted to target 92 miRNAs. Furthermore, a total of 58 out of above 92 miRNAs were predicted to target 2195 downstream genes according to the threshold of total context++ score ≤ −0.6 using TargetScan (Additional file S4).
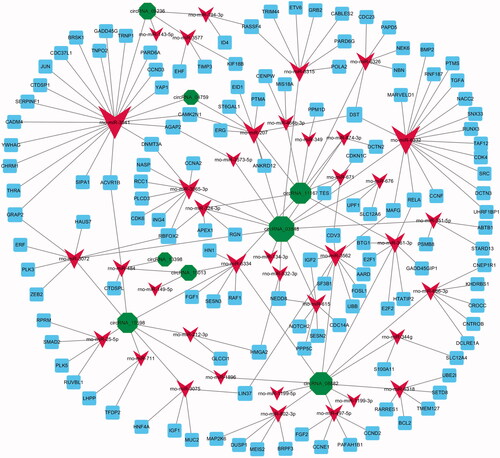
According to the threshold of total context++ score (≤ −0.6), 27 out of 35 DE circRNAs might indirectly regulate 2195 downstream mRNAs by their corresponding target miRNAs. To elucidate the potential function of 27 circRNAs during the proliferation phase of LR, we furtherly combined 2195 predicted downstream mRNAs of 58 candidate miRNAs. Finally, we established and displayed the circRNA–miRNA–mRNA networks using Cytoscape 3.2 (Additional file S5, .
Figure 3. The circRNA–miRNA–mRNA interaction networks. DE circRNAs (green node), their targeted miRNAs (red node) and genes (blue node, involved in cell proliferation) were predicted based on sequence-pairing prediction. The column size was depended on “Number of Directed Edges”.
Enrichment of DE circRNAs during the proliferation phase of rat LR
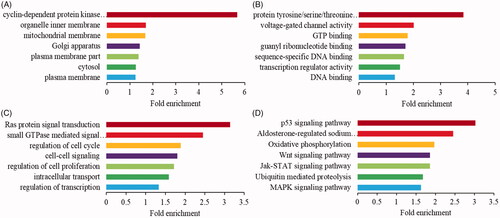
To explore the potential functions of above 27 DE circRNAs, GO and KEGG enrichment analyses were performed to classify the downstream mRNAs. The downstream mRNAs mainly encoded cellular components of the plasma membrane, organelle membrane, mitochondrial membrane, etc. () and associated with transcription regulator activity, GTP binding, DNA binding, etc. molecular functions (). Simultaneously, they were enriched in regulation of cell proliferation, small GTPase mediated signal transduction, intracellular signalling cascade, etc. biological process (). Moreover, KEGG showed that they were significantly (p ≤ .05) enriched in p53, MAPK, Wnt and Jak-STAT signalling pathway (). It predicted that most circRNAs might play a vital role in acting as a “sponge” or activating factor in organelle membrane to regulate cell proliferation through p53, MAPK, Wnt and Jak-STAT signalling pathway during the proliferation phase of rat LR.
Figure 4. Enrichment analysis of indirectly downstream mRNAs for differentially expressed circRNAs. The GO enrichment analysis of cellular component (A), molecular function (B) and biological process (C). (D) The KEGG pathway enrichment analysis.
Screening of candidate key circRNAs during the proliferation phase of rat LR
In this study, the criteria for screening the candidate key circRNAs were based on the number of the corresponding miRNA and their role enriched and reported in cell proliferation of LR or HCC (). Additionally, the expression profiles of miRNA and mRNA in the regenerating livers of rats during the proliferation phase of LR were reported in a published paper of our group (DOI:10.1002/jcp.28529). Combined with previous studies and as shown in 4 out of above 27 DE circRNAs, including circ_03848, circ_08236, circ_13398 and circ_15013 were considered as the candidate key circRNAs. For example, circ_03848 was predicted to bind to 4 miRNAs (rno-miR-207, rno-miR-30c-1-3p, rno-miR-532-3p and miR-150-5p) which were reported involved in LR. While the downstream mRNAs of these miRNAs were associated with transcription regulator activity, protein transport, Ras signal transduction and so on. Furthermore, the network of the candidate key circRNAs–miRNAs–mRNAs, which with a correlated expression pattern according and associated with cell proliferation was constructed by Cytoscape 3.2 ().
Figure 5. The circRNA–miRNA–mRNA interaction networks and expression profile of the candidate key circRNAs. (A) The candidate key circRNAs and their targeted miRNAs were displayed by green and red node, respectively. (B) DE RNA expression profile of the ceRNA network during the proliferation phase of rat LR. (C) The candidate key circRNAs, their targeted miRNAs and downstream mRNAs were displayed by green, red and blue node, respectively. The column size was depended on “Number of Directed Edges”.
Validation of the candidate key circRNA
To test whether above candidate key circRNAs were indeed circular, PCR was performed using divergent specific primers with cDNA or genomic DNA (gDNA) firstly. It showed that they could be amplified using cDNA but not gDNA (). The PCR products of these amplified circRNAs were identified directly by sequencing. The analysis of sequencing data confirmed that head-to-tail splicing existed in these circRNAs. To further verify the circular characteristics and the expression changes of these circRNAs during the proliferation phase of rat LR, they were also amplified and quantified by qRT-PCR after RNase R digestion. The expression tendencies of them were consistent with the high-throughput sequencing data ().
Figure 6. The relative level of four candidate key circRNAs during the proliferation phase of rat LR. (A) Amplification of circRNAs using outward-facing primers with cDNA but not genomic DNA (gDNA). (B) circ_03848. (C) circ_08236. (D) circ_13398. (E) circ_15013. 
 high-throughput sequencing. The infinite was represented by 9 and the infinitesimal was represented by 0.3 for the high-throughput sequencing data.
high-throughput sequencing. The infinite was represented by 9 and the infinitesimal was represented by 0.3 for the high-throughput sequencing data.
Discussion
circRNAs were a class of molecules with important biological functions, which were first reported more than 40 years ago. Recently, a large number of circRNAs have been successfully identified in different species such as archaea [Citation19], drosophila [Citation20], rat [Citation8], sheep [Citation21], pig [Citation22] and human [Citation23]. Among them, some circRNAs were found to play an important role in cell proliferation. For example, Du et al. reported that circ-Foxo3 retarded cell cycle by forming ternary complexes with p21 and CDK2 [Citation24]. It was known that the recovery of liver mass mainly resulted from hepatocyte proliferation after 2/3 PH in rats [Citation25]. Our team studied the expression changes of circRNAs during the priming phase (0–6 h after PH) of rat LR and found that 159 circRNAs changed significantly [Citation8]. After this phase, hepatocytes entered the cell cycle in the proliferation phase (12–72 h after PH). But how circRNAs work at this phase is not clear. The present study found that 164 dysregulated circRNAs could be involved in this critical step. Among them, 164 DE circRNAs originated from 150 host linear transcripts. Six circRNAs were intergenic indicating that they did not have the corresponding linear transcripts. And seven genes could produce two or three circRNAs during the proliferation phase of rat LR.
To explore the role of circRNAs in the proliferation phase of rat LR, the ceRNAs interaction network of DE circRNAs were drawn, and GO enrichment analysis of the downstream target mRNAs were performed. As a result, the biological processes were closed with tissue regeneration, regulation of cell cycle and regulation of cell proliferation, etc. It was consistent with the previous research results that quiescent hepatocytes undergo one or two rounds of replication to restore the liver mass after PH [Citation2,Citation26]. Meanwhile, KEGG analysis showed that downstream mRNAs of DE circRNAs were connected with Ras, p53, Wnt, Jak-STAT and MAPK signalling pathways, which had been acknowledged as the important signalling pathways in the proliferation phase of rat LR. For example, Kunimoto et al. reported that the novel IQGAP3/Ras/ERK-related signalling was involved in hepatocyte proliferation of LR [Citation27], indicating the critical role of Ras signalling pathway in LR. Moreover, Stepniak et al. reported that c-Jun/AP-1 promoted G1/S transition in mice hepatocytes through a p53-dependent pathway after PH [Citation28]. In addition, Wnt/β-catenin, JAK-STAT and MAPK signalling pathways have been proved to be a major regulator of regeneration [Citation29–31]. Overview, we hypothesized that DE circRNAs might regulate the expression of downstream mRNAs through their target miRNAs, thereby controlling cell proliferation through above signalling pathways in the proliferation phase of rats LR.
After having validated their expression pattern and compared the expression trends between high-throughput sequencing and qRT-PCR, a combination of target miRNA function of circRNAs must be considered, we gave the highest priority to the candidate key circRNAs, including circ_03848, circ_08236, circ_13398 and circ_15013. Because their candidate target miRNAs were involved in LR or HCC. For instance, Chaveles et al. described that miR-207 and miR-532 were significantly downregulated in rat liver after PH during the proliferation phase [Citation32,Citation33], indicating they might play a crucial role in LR. With a negative correlated expression pattern, we found that circ_03848 was upregulated sharply, and it was predicated to inhibit the activity of rno-miR-207 and rno-miR-532 to regulate cell proliferation.
It was known that circRNA played a role in post-transcriptional regulation by several ways such as miRNA sponges or protein templates [Citation14]. After predicting circRNA–miRNA pairs, the predicted target miRNAs of circ_03848, including rno-miR-150-5p and rno-miR-378a-5p were reported to have a correlated expression pattern with it and associated with LR [Citation34,Citation35], suggesting that they could be directly bound to circ_03848 to affect rat LR. In detail, it was found that circ_03848 exhibited an upregulated pattern at 30 h after PH in this paper, and it might target rno-miR-150-5p and rno-miR-378a-5p. Coincidentally, Yu et al. reported that the expression of miR-150 exhibited the sharpest downregulation from 12 to 48 h after PH during LR [Citation18]. In addition, miR-378 expression declined, accompanied by remarkably increased expression of its target ornithine decarboxylase 1 (ODC1) from 0 to 18 h after PH [Citation36], and ODC1 was found that involved in positive regulation of cell population proliferation [Citation37]. Our results were consistent with those reports by others about the expression of ceRNAs was negatively correlated [Citation38], showing that circ_03848 might target these miRNAs and affect downstream gene expression in regulating cell proliferation. Additionally, circ_08236 exhibited an upregulated pattern at 36 h after PH and predicted to target rno-miR-143-5p in this paper. With a negative correlated expression pattern, rno-miR-143-5p was reported to exhibit the sharpest downregulation from 12 to 48 h after PH during LR [Citation39]. Moreover, both of circ_13398 and circ_15013 were predicted to bind to rno-miR-484, showing this miRNA could be regulated by them. Interestingly, miR-484 was highly expressed in tumor liver compared to normal liver [Citation40]. And Yang et al. demonstrated that miR-DEN-induced precancerous lesions and HCC were dramatically impaired in miR-484−/− mice [Citation41]. Coincidentally, we also found that rno-miR-484 was upregulated in the proliferation phase of rat LR, suggesting that it might play an important role. Therefore, these candidate key circRNAs were very likely to contribute to cell proliferation by sponging their target miRNAs during the proliferation phase of rat LR.
The ceRNAs network and bioinformatics analysis preliminarily revealed the role of circRNAs in regulating cell proliferation of LR in this paper. For example, during the proliferation phase of rat LR, circ_03848 might sponge rno-miR-378a-5p and/or rno-miR-532-3p and promote the expression of their target genes ODC1 and/or RAD51, in turn, accelerate cell proliferation. In addition, circ_03848 might also be combined with rno-miR-324-3p and promote the expression of APEX1 to regulating the G1/S transformation and thus promote the process of cell proliferation. It was consistent with the results of He et al. [Citation33,Citation36,Citation42]. Additionally, Assy et al. found that the vascular endothelial growth factor A (VEGFα) played an important role in rat LR which induced by PH [Citation43]. Schug and Yu et al. found that rno-miR-143-5p and miR-150 were significantly downregulated in regenerating liver at 12–48 h after PH, and both of them regulated LR through targeting VEGFα [Citation18,Citation39]. It was consistent with the results in this study, and presumably circ_03848 and/or circ_08236 might regulate rat LR process through rno-miR-143-5p/VEGFα and/or rno-miR-150/VEGFα axis. In summary, we made scientific speculation that circ_03848, circ_08236, circ_13398 and circ_15013 regulated hepatocytes proliferation through the ceRNAs network of circRNA–miRNA–mRNA axis in the proliferation phase, thereby regulating the process of LR.
In conclusion, 10,003 circRNAs were detected and 164 of them were preliminarily determined to change remarkably in the proliferation phase of rat LR using high-throughput RNA sequencing. The ceRNAs network and bioinformatics analysis preliminarily revealed the potential role of DE circRNAs in LR, suggesting that DE circRNAs might regulate tissue regeneration and cell proliferation through Ras, p53, Wnt, Jak-STAT and MAPK signalling pathways during the proliferation phase of LR. And then, four kinds of circRNAs, including circ_03848, circ_08236, circ_13398 and circ_15013 were considered as the candidate key circRNAs and speculated to regulate cell proliferation at 12–72 h after PH by sponging miRNAs and modulating the downstream mRNAs, which based on the number of the corresponding miRNAs and their role enriched and reported in cell proliferation of LR or HCC. It will provide the novel information for LR. In the further studies, using gene interference or additive techniques to verify our conclusion that circRNAs regulate the process of LR are necessary.
Supplementary_date.rar
Download ()Disclosure statement
The authors report no conflict of interest
Additional information
Funding
Related Research Data
References
- Zimmermann A. Regulation of liver regeneration. Nephrol Dial Transplant. 2004;19(4):iv6–10.
- Michalopoulos GK, DeFrances M. Liver regeneration. Adv Biochem Eng Biotechnol. 2005;93:101–134.
- Yuceturk H, Yagmurdur MC, Gur G, et al. Role of heparin on TNF-alpha and IL-6 levels in liver regeneration after partial hepatic resection. Eur Surg Res. 2007;39:216–221.
- Adamek B, Zalewska-Ziob M, Strzelczyk JK, et al. Hepatocyte growth factor and epidermal growth factor activity during later stages of rat liver regeneration upon interferon alpha-2b influence. Clin Exp Hepatol. 2017;3:9–15.
- Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013;495:333–338.
- Fanale D, Taverna S, Russo A, et al. Circular RNA in Exosomes. Adv Exp Med Biol. 2018;1087:109–117.
- Xu Z, Li P, Fan L, et al. The potential role of circRNA in tumor immunity regulation and immunotherapy. Front Immunol. 2018;9:9.
- Li L, Guo J, Chen Y, et al. Comprehensive circRNA expression profile and selection of key circRNAs during priming phase of rat liver regeneration. BMC Genomics. 2017;18:80.
- Higgins GA. Experimental pathology of the liver: restoration of the liver of the white rat following partial surgical removal. Arch Pathol 1931;12:186–202.
- John B, Enright AJ, Aravin A, et al. Human MicroRNA targets. PLoS Biol. 2004;2:e363.
- Fromm B, Billipp T, Peck LE, et al. A uniform system for the annotation of vertebrate microRNA genes and the evolution of the human microRNAome. Annu Rev Genet. 2015;49:213–242.
- Shi Y, Yang F, Wei S, et al. Identification of key genes affecting results of hyperthermia in osteosarcoma based on integrative ChIP-Seq/TargetScan analysis. Med Sci Monit. 2017;23:2042–2048.
- Huang DW, Sherman BT, Tan Q, et al. DAVID bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–W175.
- Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature 2013;495:384–388.
- Inoue K. [MicroRNA function in animal development]. Tanpakushitsu Kakusan Koso. 2007;52:197–204. Japanese.
- Motawi TK, Mohamed MR, Shahin NN, et al. Time-course expression profile and diagnostic potential of a miRNA panel in exosomes and total serum in acute liver injury. Int J Biochem Cell Biol. 2018;100:11–21.
- Lu J, Chen C, Hao L, et al. MiRNA expression profile of ionizing radiation-induced liver injury in mouse using deep sequencing. Cell Biol Int. 2016;40:873–886.
- Yu ZY, Bai YN, Luo LX, et al. Expression of microRNA-150 targeting vascular endothelial growth factor-A is downregulated under hypoxia during liver regeneration. Mol Med Rep. 2013;8:287–293.
- Danan M, Schwartz S, Edelheit S, et al. Transcriptome-wide discovery of circular RNAs in Archaea. Nucleic Acids Res. 2012;40:3131–3142.
- Kramer MC, Liang D, Tatomer DC, et al. Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins. Genes Dev. 2015;29:2168–2182.
- Cao Y, You S, Yao Y, et al. Expression profiles of circular RNAs in sheep skeletal muscle. Asian-Australas J Anim Sci. 2018;31:1550–1557.
- Huang M, Shen Y, Mao H, et al. Circular RNA expression profiles in the porcine liver of two distinct phenotype pig breeds. Asian-Australas J Anim Sci. 2018;31:812–819.
- Werfel S, Nothjunge S, Schwarzmayr T, et al. Characterization of circular RNAs in human, mouse and rat hearts. J Mol Cell Cardiol. 2016;98:103–107.
- Du WW, Yang W, Liu E, et al. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016;44:2846–2858.
- Michalopoulos GK. Advances in liver regeneration. Expert Rev Gastroenterol Hepatol. 2014;8:897–907.
- Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43:S45–S53.
- Kunimoto K, Nojima H, Yamazaki Y, et al. Involvement of IQGAP3, a regulator of Ras/ERK-related cascade, in hepatocyte proliferation in mouse liver regeneration and development. J Cell Physiol. 2009;220:621–631.
- Stepniak E, Ricci R, Eferl R, et al. c-Jun/AP-1 controls liver regeneration by repressing p53/p21 and p38 MAPK activity. Genes Dev. 2006;20:2306–2314.
- Phesse TJ, Buchert M, Stuart E, et al. Partial inhibition of gp130-Jak-Stat3 signaling prevents Wnt-beta-catenin-mediated intestinal tumor growth and regeneration. Sci Signal. 2014;7:ra92.
- Ozaki M. [Role of jak/STAT3 and PI3-K/Akt pathways in liver injury and regeneration]. Seikagaku. 2008;80:399–408. Japanese.
- Lee SC, Jeong HJ, Lee SK, et al. Hypoxic conditioned medium from human adipose-derived stem cells promotes mouse liver regeneration through JAK/STAT3 signaling. Stem Cells Transl Med. 2016;5:816–825.
- Chaveles I, Zaravinos A, Habeos IG, et al. MicroRNA profiling in murine liver after partial hepatectomy. Int J Mol Med. 2012;29:747–755.
- Raschzok N, Werner W, Sallmon H, et al. Temporal expression profiles indicate a primary function for microRNA during the peak of DNA replication after rat partial hepatectomy. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1363–R1372.
- Geng X, Chang C, Zang X, et al. Integrative proteomic and microRNA analysis of the priming phase during rat liver regeneration. Gene 2016;575:224–232.
- Kim J, Hyun J, Wang S, et al. MicroRNA-378 is involved in hedgehog-driven epithelial-to-mesenchymal transition in hepatocytes of regenerating liver. Cell Death Dis. 2018;9:721.
- Song G, Sharma AD, Roll GR, et al. MicroRNAs control hepatocyte proliferation during liver regeneration. Hepatology. 2010;51:1735–1743.
- Lee NK, Skinner JP, Zajac JD, et al. Ornithine decarboxylase is upregulated by the androgen receptor in skeletal muscle and regulates myoblast proliferation. Am J Physiol Endocrinol Metab. 2011;301:E172–E179.
- Sang Y, Chen B, Song X, et al. circRNA_0025202 regulates tamoxifen sensitivity and tumor progression via regulating the miR-182-5p/FOXO3a axis in breast cancer. Mol Ther. 2019. [Epub ahead of print].
- Schug J, McKenna LB, Walton G, et al. Dynamic recruitment of microRNAs to their mRNA targets in the regenerating liver. BMC Genomics. 2013;14:264.
- Tessitore A, Cicciarelli G, Del Vecchio F, et al. MicroRNA expression analysis in high fat diet-induced NAFLD-NASH-HCC progression: study on C57BL/6J mice. BMC Cancer. 2016;16:3.
- Yang Y, Lin X, Lu X, et al. Interferon-microRNA signalling drives liver precancerous lesion formation and hepatocarcinogenesis. Gut 2016;65:1186–1201.
- He T, Weintraub NL, Goswami PC, et al. Redox factor-1 contributes to the regulation of progression from G0/G1 to S by PDGF in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2003;285:H804–H812.
- Assy N, Spira G, Paizi M, et al. Effect of vascular endothelial growth factor on hepatic regenerative activity following partial hepatectomy in rats. J Hepatol. 1999;30:911–915.