
Abstract
Cervical spondylosis is a degenerative disease commonly found in older adults and characterized by progressive osteophyte formation and disc collapse. Apoptosis in nucleus pulposus (NP) cells which induced by TNF-α has been widely known to associate with the disc degeneration. However, the exactly underlying molecular mechanism was still unclear. The aim of our study was to investigate whether TRIM14/NF-κB signalling pathway is associated with the apoptosis of human NP cells (HNPC) induced by TNF-α. Our data demonstrated that TNF-α treatment obviously decreased the cell viability, induced apoptosis and increased TRIM14 expression and NF-κBp65 activity in HNPC in a dose-dependent manner. Down-regulation of TRIM14 or NF-κB inhibitor PDTC treatment significantly inhibited cell apoptosis, Bax/Bcl-2 ratio and NF-κBp65 activation induced by TNF-α in HNPC. Meanwhile, up-regulation of TRIM14 obviously increased cell apoptosis, Bax/Bcl-2 ratio and NF-κBp65 activation in HNPC. Then, we found that the protein PPM1A was identified as a binding partner of TRIM14 and ubiquitinated by TRIM14. These findings provide insights into the function of TRIM14 and NF-κB signalling and might, therefore, represent a novel therapeutic target for treatment of cervical spondylosis.
Introduction
Cervical spondylosis (CS) is caused by natural ageing and the unspecified degenerative changes in the neck and shoulder bones, muscles, tendons and joints [Citation1]. Among them, the degeneration of intervertebral disc is especially important because it is the first factor of the structural degeneration of the cervical spine and has evolved a series of pathological and pathophysiological changes of the CS [Citation2]. Degenerative changes begin with osteophyte formation in the intervertebral disc and cause a series of physiological changes that lead to biomechanical changes that cause nerve and vascular compression [Citation3]. However, additional disc damage also increases the degenerative process in young patients. CS often causes neck stiffness, pain and other symptom, and pain and numbness are sometimes appeared in arms and at the root of the fingers [Citation4,Citation5]. Epidemiological studies have found that up to 86.6% of CS patients suffer from neck pain [Citation6]. Another type of CS is associated with vascular and nerve compression, which is caused by narrowing of the intervertebral space and collision between osteophyte formation, then appears numb, shock and pain, and if not corrected will cause permanent disability. Drug treatment of CS mainly through the promotion of neck movement and reduce the degree of pain. Long term complementary treatments include acupuncture, neck exercise, mechanical traction, massage and electrotherapy, which can relieve neck pain in patients with CS [Citation7,Citation8]. However, these therapies have not been widely used because of the lack of sufficient and accurate evidence to reveal their safety and efficacy.
Cell density reduction and catabolism of extracellular matrix is of great importance in intervertebral disc degeneration induced by apoptosis of intervertebral discs [Citation9]. Degenerative disc disease is characterized by homeostatic regulation of disc degeneration, leading to programmed cell death of the nucleus pulposus. Nucleus pulposus is a part of the structure of intervertebral disc, located between the two cartilage plate and the fibrous ring, which is composed of typical small cartilage-like cells, and the phylogenetic profiles between nucleus pulposus and cartilage were similar at the molecular level [Citation10]. Among a large number of cytokines, TNF-α is thought to be associated with disc degeneration, since it may enhance the expression of matrix-degrading enzymes which are responsible for cleaving collagen type II as well as proteoglycan [Citation11], and is of great importance in the development and progression of disc degeneration [Citation12,Citation13]. In the course of disc degeneration, in addition to infiltrating immune cells, the innate nucleus pulposus cells also produce high levels of cytokines, including TNF-α [Citation14]. Therefore, it is very important for the development of this strategy to clarify the mechanism of apoptosis of intervertebral disc cells.
Tripartite motif-containing (TRIM) family proteins are associated with various biological processes, including cell differentiation, apoptosis, regulation of cell cycle and transcription, innate immunity and antiviral activity [Citation15,Citation16]. In recent years, TRIM14 has been used as a mitochondrial adaptor to regulate innate immune responses by linking mitochondrial antiviral signaling protein (MAVS) and NF-κB essential and modulator (NEMO) [Citation17]. TRIM14 can inhibit replication of vesicular stomatitis virus by enhancing IFN-α pathway and NF-κB production, but mutant TRIM14 is not. However, the replication and infection of HCV by TRIM14 modulation in JFH and Huh7 cells does not depend on the up-regulation of IFN-α pathway and NF-κB production [Citation18]. Enhanced TRIM14 expression can reduce the replication of sindbis virus in HEK293 cells and simultaneously activate transcription factor NF-κB subunits, thus participating in innate immunity [Citation19]. Moreover, increased expression of TRIM14 activates the NF-κB signaling pathway and is involved in the aggressiveness process in tongue squamous cell carcinoma [Citation20]. Thus, these studies suggest that enhanced TRIM14 is closely related to the activation of the NF-κB pathway.
Although an increasing body of evidence has indicated the great importance of NF-κB signalling pathway in the cell apoptosis of NP cells, the role of TRIM14/NF-κB signalling in cell apoptosis of degenerative HNPC induced by TNF-α has not been previously investigated. Our study provides a further exploration focused on whether TNF-α induces apoptosis of HNPC via activating the TRIM14/NF-κB signalling pathway.
Materials and methods
Clinical sample collection
Primary human nucleus pulposus tissues were isolated from NP of human intervertebral disc of 15 normal subjects and 30 patients of cervical spondylosis undergoing surgery for microdiscectomy or discectomy and fusion in The Affiliated Shanghai General Hospital of Nanjing Medical University. The informed consent from all the participants was obtained. This prospective study was approved by the Ethics Committee of The Affiliated Shanghai General Hospital of Nanjing Medical University. The nucleus pulposus tissues were collected in culture medium, washed with phosphate buffered saline under sterile conditions and used for Real-time PCR assay.
HNPC culture and treatment
HNPC were cultured in DMEM with high glucose (Hyclone; GE Healthcare Life Sciences, Logan, UT, USA), commonly with 10% FBS and 1% penicillin-streptomycin mixture (Gibco BRL, Grand Island, NY, USA) supplementation, at 37 °C under a humidified atmosphere with 5% CO2. HNPC were treated with either different doses of TNF-α or appropriate concentration of TNF-α with 10 µmol/l PDTC treatment or pLKO.1-shRNA-TRIM14 (shTRIM14) transfection or with pLVX-Puro-TRIM14 transfection individually.
Lentiviral transduction
RNA interference (RNAi) constructs with sequences targeting human TRIM14 (point 1676–1698 shRNA-1, 5′-ACAGGCCCTAAACAAGTCT-3′, point 1864–1886 shRNA-2, 5′-GTAGGCACACAGTAAACGT-3′, and point 3114–3136 shRNA-3, 5′-TAGACTTTCAGTGCATTAGT-3′) or scramble shRNA negative control were cloned into the pLKO.1 vector. The scramble shRNA cloned into the pLKO.1 vector was used as a negative control. The constructs containing pLKO.1-shRNA-TRIM14 (shTRIM14), psPAX2 and pMD2G were then co-transfected into HEK 293 T cells for 4–6 h using Lipofectamine 2000 reagent following the manufacturer’s protocol. Subsequently, 48 h after transfection, particles in cell culture medium were collected and used to transduce HNPC infection.
The cDNA encoding TRIM14 was cloned into the pLVX-Puro for generating TRIM14 expression vector. The forward primer was 5′-GCGAATTCATGGCGGGCGCGGCGACC-3′ and the reverse primer was 5′-CGGGATCCCTAGGGCAGCCGGGGGAT-3′. The pLVX-Puro-TRIM14, psPAX2 and pMD2G were then co-transfected into the 293 T cells for 4–6 h using Lipofectamine 2000 reagent following the manufacturer’s protocol. HNPC were then infected with pLVX-Puro-TRIM14. The blank vector of pLVX-Puro was used as negative control.
Cell viability assay
The cell viability of HNPC was measured by CCK-8 assay. Briefly, the HNPC (3 × 103 cells/well) were cultured with 5% CO2 at 37 °C overnight. After treatment with different doses of TNF-α (0, 30, 50 and 100 ng/ml) for 0, 24, 48 and 72 h, the culture media were replaced with 10% CCK-8 solution and cells were cultured at 37 °C for 1 h. Optical density at wavelength 450 nm (OD450) was measured using a VERSAmax tunable microplate reader (Sunnyvale, CA, USA).
Flow cytometry assay
The apoptotic rate of HNPC was measured by flow cytometry assay. In brief, HNPC were seeded at a density of 2 × 105 cells/ml and washed with 1 ml Binding Buffer (3 ml 10× Binding Buffer in 27 ml ddH2O). Subsequently, the cells were stained with 195 µl Annexin V-FITC for 10 min in dark room and with 5 µl Propidium Iodide (PI) for 5 min. Apoptotic cells were analyzed by Accuri C6 Flow Cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Quantitative real-time RT-PCR
Total RNA from HNPC or primary human nucleus pulposus tissues was extracted with Trizol Reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instruction, and iScript TM cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA, USA) was used to carry out reverse transcription reaction on RNA. Real-time PCR was performed using SYBR Green (Takara, Otsu, Shiga, Japan) and the GeneAmp PCR Systems 2700 (Applied Biosystems). The primer sequences of the genes used in quantitative PCR were shown subsequently: TRIM14 forward, 5′-GGATTTGTGTCTCCGTTCTG-3′; TRIM14 reverse, 5′-TCTGTCTGCCTGGTATTCTG-3′; PPM1A forward, 5′-ATGAGTCAGCACTGCCATAC-3′; PPM1A reverse, 5′-CCCACTATTACCTCCAGGAATC-3′; TNF-α forward, 5′-ACGGAGCTGAACAATAGG-3′; TNF-α reverse, 5′-GGCGATTACAGACACAAC-3′; NF-κBp65 forward, 5′-GAATGGCTCGTCTGTAGTG-3′; NF-κBp65 reverse, 5′-TGGTATCTGTGCTCCTCTC-3′; GAPDH forward, 5′-CACCCACTCCTCCACCTTTG-3′; as well as GAPDH reverse, 5′-CCACCACCCTGTTGCTGTAG-3′. The relative fold changes in mRNA expression were calculated using the 2−ΔΔCt method.
Western blotting
Total protein was extracted from HNPC the addition of RIPA lysis buffer supplemented with protease inhibitor cocktail (P‐8340; Sigma, St Louis, MO, USA). Total protein (25 μg) got divided by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and was subsequently transferred to a nitrocellulose membrane (Whatman®, GE Healthcare, Dassel, Germany), which was then immunoblotted at 4 °C for 12 h with anti-TRIM14 (Abcam, Cambridge, MA, USA; ab185349), anti-NF-κBp65 (Cell Signaling Technology, Danvers, MA, USA; #8242), anti-p-NF-κBp65 (Cell Signaling Technology; #3033), anti-Bax (Santa Cruz Biotech., Santa Cruz, CA, USA; sc-493), anti-Bcl-2 (Santa Cruz Biotech.; sc-492), anti-PPM1A (Cell Signaling Technology; #3549S), anti-PPM1B (Abcam; ab70804) and anti-GAPDH (Cell Signaling Technology; #5174). Horseradish peroxidase-conjugated secondary antibodies (1:1000; Beyotime Institute of Biotechnology, Shanghai, China; A0208, A0181 and A0216) were used to incubate membranes for one hour at 37 °C. Finally, the membranes were washed and subsequently incubated with Enhanced chemiluminescence (ECL) detection kit (Amersham Bioscience, Amersham, UK) for image scanning. The density of each protein band was analyzed using LAS 3000 imaging system (Fujifilm, Tokyo, Japan).
Co-immunoprecipitation and ubiquitination
Co-immunoprecipitation and ubiquitination were performed as described previously [Citation21,Citation22]. Briefly, cells infected with shTRIM14 in the presence of TNF-α were lysed with RIPA buffer, reacted with normal IgG (Santa Cruz Biotech.; sc-2027) or anti-PPM1A (Cell Signaling Technology; #3549S) antibody overnight at 4 °C and then with protein A/G Plus agarose (Santa Cruz Biotech.; sc-2003) for 2 h at 4 °C. The immunoprecipitated complexes were washed three times in lysis buffer and analyzed by Western blot analysis using anti-PPM1A, anti-TRIMM14 or anti-Ubiquitin (Abcam; ab7780) antibody.
Statistical analysis
Results are expressed as mean ± the standard deviation from 3 independent experiments and evaluated with software of SPSS (Version 18.0, IBM Corp., Armonk, NY, USA). Significance of the differences was determined using ANOVA or unpaired, two-tailed Student’s t-tests. When p was less than .05, it had statistical significance.
Results
TNF-α inhibits cell viability and induces apoptosis of HNPC
To evaluate the function of TNF-α in regulating the viability and apoptosis of HNPC, HNPC were treated with different concentrations of TNF-α (30, 50 and 100 ng/ml) for indicated time, and CCK-8, as well as flow cytometry assay, was performed, respectively. Our results showed that TNF-α (30, 50 and 100 ng/ml) treatment markedly inhibited cell viability of HNPC in dose- and time-dependent manner compared with control (). After 100 ng/ml of TNF-α treatment for 24, 48 and 72 h, the cell viability of HNPC was decreased by 39.5%, 46.4% and 60.9% compared with control, respectively. Moreover, TNF-α (30, 50 and 100 ng/ml) treatment for 24 h also significantly increased cell apoptosis of HNPC by 11.6-fold, 22.9-fold and 34.4-fold compared with control, respectively ().
Figure 1. TNF-α inhibited cell viability and induced apoptosis of HNPC. HNPC were treated with different concentrations of TNF-α (30, 50 and 100 ng/ml), and the cell viability (A) and apoptosis (B,C) was measured using CCK-8 and flow cytometry assay, respectively. **p < .01 compared with control.
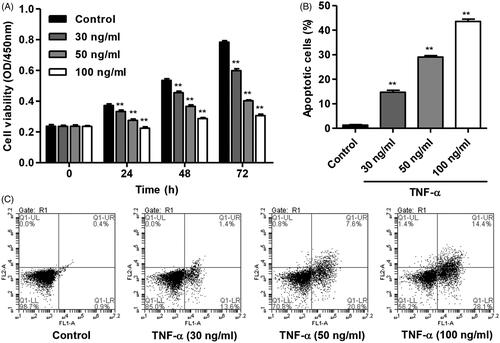
TNF-α increases TRIM14 and NF-κBp65 expression in HNPC
To investigate the effect of TNF-α on TRIM14 expression and NF-κB activation in HNPC, the expression of TRIM14 and NF-κBp65 was measured by Western blot and Real-time PCR assay, respectively. As shown in , TNF-α (30, 50 and 100 ng/ml) treatment for 24 h obviously increased the mRNA levels of TRIM14 and NF-κBp65 in HNPC in a dose-dependent manner compared with control. After 100 ng/ml of TNF-α treatment, the mRNA levels of TRIM14 and NF-κBp65 was increased by 4.8-fold and 4.5-fold, respectively. The similar results were also found in the protein levels of TRIM14 and NF-κBp65 in HNPC (). After 100 ng/ml of TNF-α treatment the mRNA levels of TRIM14 and NF-κBp65 was increased by 2.6-fold and 3.3-fold compared with control, respectively. 100 ng/ml of TNF-α was therefore used for subsequent experiments.
Figure 2. TNF-α increased TRIM14 and NF-κBp65 expression of HNPC. HNPC were treated with different concentrations of TNF-α (30, 50 and 100 ng/ml), and the expression of TRIM14 and NF-κBp65 was measured using Western blot and Real-time PCR assay, respectively. **p < .01 compared with control.
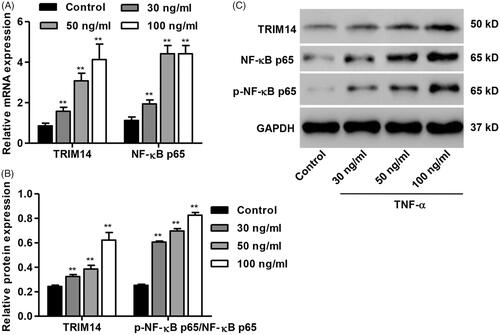
Down-regulation of TRIM14 inhibits apoptosis and NF-κBp65 expression in HNPC induced by TNF-α
To further study the role of TRIM14 and NF-κBp65 signalling in TNF-α-induced HNPC apoptosis, TRIM14 was down-regulated in HNPC by transfection with pLKO.1-shRNA-TRIM14 (shTRIM14), and the NF-κB inhibitor, PDTC, was also introduced. As shown in , HNPC were respectively transfected with sh-TRIM14-1, sh-TRIM14-2 or sh-TRIM14-3 and showed significantly decreased TRIM14 mRNA expression by 89.5%, 83.8% and 84.0% compared with control, respectively. sh-TRIM14-1 was therefore used for subsequent experiments. As we expected, TNF-α treatment for 24 h significantly increased cell apoptosis of HNPC compared with control, but was reversed by TRIM14 down-regulation as well as 10 µmol/L PDTC treatment (). Moreover, TNF-α treatment induced TRIM14 expression and NF-κBp65 activation and increased the ratio of Bax/Bcl-2 protein expression by 4.6-fold (). However, these protein levels were significantly decreased by TRIM14 down-regulation or PDTC treatment.
Figure 3. TRIM14 down-regulation inhibited apoptosis and related protein expression in HNPC induced by TNF-α. HNPC were treated with 100 ng/ml of TNF-α with or without pLKO.1-shRNA-TRIM14 (shTRIM14) and PDTC treatment. (A) TRIM14 mRNA expression was determined using Real-time PCR assay. (B,C) HNPC apoptosis was determined using flow cytometry assay. (D,E) The level of TRIM14, NF-κBp65, p-NF-κBp65, Bax and Bcl-2 protein was measured by Western blot assay. **p < .01 compared with control. ##p < .01 compared with TNF-α treatment.
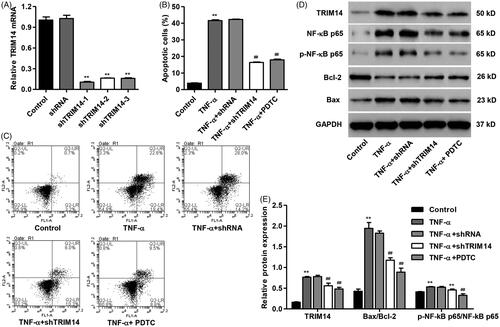
Up-regulation of TRIM14 promotes apoptosis and NF-κBp65 expression in HNPC
To assess the function of TRIM14 in HNPC apoptosis and its underlying molecular mechanism, HNPC was transfected with pLVX-Puro-TRIM14 expressing vector responsible for overexpression of TRIM14. Our data showed that overexpression of TRIM14 markedly increased the mRNA level of TRIM14 by 8.7-fold () and induced HNPC apoptosis by 25.9-fold compared with control (). In addition, overexpression of TRIM14 also increased the protein levels of TRIM14 and Bax/Bcl-2 ratio and NF-κBp65 activation by 2.5-fold, 2.3-fold and 5.4-fold compared with control, respectively ().
Figure 4. TRIM14 up-regulation induced apoptosis and related protein expression in HNPC. HNPC were transfected with pLVX-Puro-TRIM14 expressing vector. (A) TRIM14 mRNA expression was determined using Real-time PCR assay. (B,C) HNPC apoptosis was determined using flow cytometry assay. (D,E) The level of TRIM14, NF-κBp65, p-NF-κBp65, Bax and Bcl-2 protein was determined using Western blot assay. **p < .01 compared with control.
Association of TRIM14 with PPM1A in HNPC
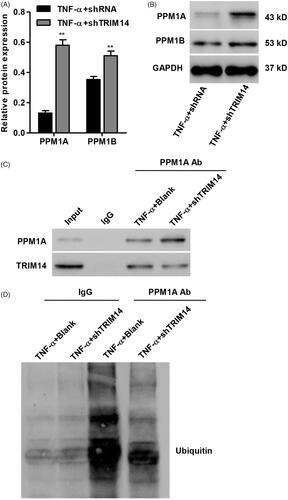
To determine the possible molecular mechanism through which TRIM14 regulates TNF-α-induced apoptosis in HNPC, we identified protein candidates that functionally associated with TRIM14. As shown in , the protein expression of PPM1A and PPM1B was significantly increased in TNF-α induced HNPC with TRIM14 down-regulation, with more increased protein expression detected in PPM1A. PPM1A was therefore used for following co-immunoprecipitiation assay. Our resulted showed that PPM1A directly interacts with TRIM14 in HNPC (). Moreover, ubiquitinated PPM1A was detected using an anti-ubiquitin antibody. showed that down-regulation of TRIM14 markedly suppressed PPM1A ubiquitination in HNPC with TNF-α treatment.
Figure 5. TRIM14 binds to and ubiquitination of PPM1A in vitro. (A,B) The protein expression of PPM1A and PPM1B in TNF-α-induced HNPC was measured by Western blot assay. (C) Co-immunoprecipitation showed that TRIM14 interacts with PPM1A in TNF-α-induced HNPC with TRIM14 down-regulation. (D) HNPC with TRIM14 down-regulation was treated with TNF-α, and PPM1A was immunoprecipitated and immunoblotted. **p < .01 compared with control.
Expression of TRIM14, PPM1A, TNF-α and NF-κBp65 in patients with cervical spondylosis
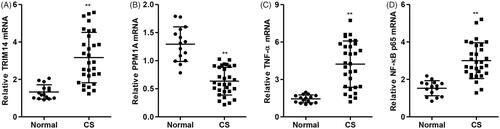
We then test the mRNA expression of TRIM14, PPM1A, TNF-α and NF-κBp65 in patients with cervical spondylosis, and the results demonstrated that the mRNA expression of TRIM14, TNF-α and NF-κBp65 was significantly increased, but PPM1A mRNA expression was decreased in primary human nucleus pulposus tissues from patients with cervical spondylosis compared with healthy controls ().
Figure 6. TRIM14, PPM1A, TNF-α and NF-κBp65 mRNA expression in patients with cervical spondylosis. The mRNA expression of TRIM14 (A), PPM1A (B), TNF-α (C) and NF-κBp65 (D) expression in primary human nucleus pulposus tissues in patients with cervical spondylosis (n = 30) and normal subject controls (n = 15) was measured by Real-time PCR. Data are expressed as mean ± SD. **p < .01 compared with control.
Discussion
Cervical spondylosis is a common progressive degenerative disease of the human spinal column that occurs more than 50 years of age and widely presents as myelopathy or radiculopathy [Citation23]. Early cervical spondylosis is associated with degenerative changes caused by disc dryness, resulting in narrowing of the intervertebral space, while reducing the ability of the intervertebral disc to maintain or sustain additional loads from the cervical axis [Citation1]. Disc degeneration is associated with extracellular matrix degradation and increased apoptosis of nucleus pulposus cells [Citation24]. Therefore, searching for factors that can suppress stress-induced apoptosis of nucleus pulposus cells can provide important strategies for the treatment of CS. In the present study, our data demonstrated that TNF-α treatment markedly inhibited cell viability and induced apoptosis in HNPC as well as increased the expression of TRIM14 and NF-κBp65 activation. Overexpression of TRIM14 mimics the effects of TNF-α on cell viability and apoptosis in HNPC, while down-regulation of TRIM14 by shRNA obviously inhibited TNF-α induced cell viability and apoptosis in HNPC. Importantly, TRIM14 directly interacts with PPM1A and ubiquitination of PPM1A in TNF-α induced HNPC. Moreover, TRIM14, TNF-α and NF-κBp65 expression were increased in patients with CS, but PPM1A was decreased.
In particular, dysfunction of apoptosis and increased speed of ageing, which are the main cellular processes in NP cells and play an essential role in disc degeneration [Citation25,Citation26]. Nucleus pulposus cells, known as type 2 apoptotic cells, rely on two independent but related pathways: intrinsic mitochondrial pathways and extrinsic pathways that depend on the Fas receptor. Apoptosis of nucleus pulposus caused by various pathways, including abnormal mechanical pressure, serum clearance, oxidative stress, cytokines and nitric oxide, is of great importance in the pathological progression of human intervertebral disc degeneration in vivo and in vitro [Citation27,Citation28]. TNF-α plays an important role in the development of intervertebral disc degeneration and is the most commonly used drug in the study of intervertebral disc degeneration in vitro. Excessive apoptosis can accelerate the progression of degeneration of intervertebral disc so that the inhibition of apoptosis can effectively alleviate the degeneration of intervertebral disc [Citation29]. In this study, we found that TNF-α decreased cell viability in time- and dose-dependent manner. Meanwhile, TNF-α treatment markedly induced apoptosis and increased the expression of TRIM14 and NF-κBp65 activation in HNPC in a dose-dependent manner.
Activation of NF-κB is a strictly regulated process. In normal cells, NF-κB is activated only when stimulated properly and positively regulates transcription of target genes. NF-κB can be activated in a few minutes by a variety of stimuli, including immune cytokines such as TNF-α and IL-1, T-cell activation signals, growth factors, and stress inducers [Citation30], suggesting that activation of NF-κB is an inducible transient process. In addition, activation of other signalling pathways such as Ras/MAPK and PI3K/Akt also activates NF-κB [Citation31]. Classical NF-κB activation pathways triggered by immune stimulating factors such as TNF-α and IL-1β have been extensively studied. NF-κB is a pleiotropic transcription factor involved in the induction of multiple gene expression. It is found that NF-κB can be activated during the prophase of apoptosis or in the process of apoptosis by stimulating factors, speculated that NF-κB activation is closely related to the apoptosis [Citation32,Citation33]. Consistent with the previous studies, our results also demonstrated that PDTC, the NF-κB inhibitor, not only significantly inhibited activation of NF-κBp65, but also inhibited cell apoptosis and the expression of TRIM14 and Bax/Bcl-2 ratio in HNPC induced by TNF-α. However, NF-κB may also activate transcription of some genes involved in the inhibition of cell death by two pathways, including the mitochondrial pathway and the death receptor pathway. NF-κB may interfere with the transcriptional activity of p53 and inhibit the apoptosis induced by p53 by regulating the expression of anti-apoptotic genes and down-regulating the expression of p53 [Citation31]. These suggested that NF-κB may have dual functions, can be used both as inhibitors of apoptosis and as activator of apoptosis, which depends on the different members of the NF-κB regulation of different signals, and the relationship between NF-κB and apoptosis also depends on the activation of signaling pathways in different cell types and different stimuli [Citation34,Citation35].
To date, very less is known about the biological and molecular mechanisms mediated by TRIM14 in either normal or pathogenic states. In the present study, our data demonstrated that down-regulation of TRIM14 significantly inhibited cell apoptosis and Bax/Bcl-2 ratio as well as the activation of NF-κBp65 in HNPC induced by TNF-α, which was corrected by up-regulation of TRIM14 in HNPC without TNF-α stimulation. Consistent with our findings, recently study found that TRIM14 down-regulation markedly promoted cell growth and soft agar colony formation and reduced cell apoptosis of non-small cell lung cancer cell lines in vitro [Citation36], suggesting an pro-apoptotic role of TRIM14 in non-small cell lung cancer cell lines. It also has been reported that TRIM14 was closely related to activation of the NF-κB pathway [Citation20]. Importantly, our data also showed that PPM1A directly interacts with TRIM14 in HNPC, and down-regulation of TRIM14 markedly suppressed PPM1A ubiquitination. PPM (protein phosphatase magnesium-dependent) enzymes is a subfamily of PTPs (protein tyrosine phosphatases) that acts on serine/threonine residues and activation required for Mg2+ or Mn2+ ions [Citation37]. PPM includes PP2C (protein phosphatase type 2 C), including PPM1A (PP2Cα) and PPM1B (PP2Cβ), and pyruvate dehydrogenase phosphatase. PPM1A and PPM1B are the most abundant and most studied PP2C isozymes. PP2C can affect different physiological processes, including stress response, metabolism and cell cycle, by binding to a variety of substrates, including kinases, receptors, channels and transcription factors [Citation38,Citation39]. Dephosphorylation of Bad can induce apoptosis, whereas PPM1A and PPM1B cause the phosphorylation of Bad, which suggested that activation of PPM1A and PPM1B may induce apoptosis [Citation40]. These results indicated that TRIM14 directly interacted with PPM1A to promote PPM1A ubiquitination and degradation, which may contribute to the apoptosis in HNPC induced by TNF-α. However, the precise molecular mechanism underlying the enhancement of PPM1A ubiquitination by TRIM14 remains elusive and warrants further studies.
In conclusion, the current study demonstrated that TNF-α treatment inhibited cell viability and induced apoptosis in HNPC, which was corrected by down-regulation of TRIM14, and similar with the effect of TRIM14 up-regulation in HNPC without TNF-α treatment. Notably, TRIM14 directly interacted with PPM1A to promote PPM1A ubiquitination and activation of NF-κBp65 was associated with the apoptosis-inducing effect of TNF-α, may therefore provide a therapeutic target for the treatment of degenerative disc disease.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Ferrara LA. The biomechanics of cervical spondylosis. Adv Orthop. 2012;2012:493605.
- Ando T. Diagnosis and management of cervical spondylosis. Rinsho Shinkeigaku. 2012;52:469–479.
- Singh M, Khurana I, Kundu ZS, et al. Link of sympathetic activity with cardiovascular risk in patients of cervical spondylosis. Int J Clin Exp Physiol. 2016;3:41.
- Piao Y. Comment on the “Assessment of a traditional acupuncture therapy for chronic neck pain: a pilot randomized controlled study”. Complement Ther Med. 2011;19:349–350.
- Hoy DG, Protani M, De R, et al. The epidemiology of neck pain. Best Pract Res Clin Rheumatol. 2010;24:783–792.
- Borenstein DG. Chronic neck pain: how to approach treatment. Curr Pain Headache Rep. 2007;11:436–439.
- Gu K, Yan Y, Wei W, et al. Safety and efficacy study of an ozone laser combined therapy using puncture needle in the treatment of patients with cervical spondylosis. Clin Spine Surg. 2017;30:E505–E509.
- Wang CM, Wang YW, Chu JM, et al. Comparison of the efficacies of transverse needling versus perpendicular needling at myofascial trigger points in treating cervical spondylosis. Shanghai J Acupunct Moxibust. 2016;25:261–264.
- Wang T, Yang SD, Liu S, et al. 17Beta-estradiol inhibites tumor necrosis factor-alpha induced apoptosis of human nucleus pulposus cells via the PI3K/Akt pathway. Med Sci Monit. 2016 22: 4312–4322.
- Hunter CJ, Matyas JR, Duncan NA. Cytomorphology of notochordal and chondrocytic cells from the nucleus pulposus: a species comparison. J Anat. 2004;205:357–362.
- Le Maitre CL, Freemont AJ, Hoyland JA. The role of interleukin-1 in the pathogenesis of human intervertebral disc degeneration. Arthritis Res Ther. 2005;7:R732–R745.
- Purmessur D, Walter BA, Roughley PJ, et al. A role for TNFalpha in intervertebral disc degeneration: a non-recoverable catabolic shift. Biochem Biophys Res Commun. 2013;433:151–156.
- Le Maitre CL, Hoyland JA, Freemont AJ. Catabolic cytokine expression in degenerate and herniated human intervertebral discs: IL-1beta and TNFalpha expression profile. Arthritis Res Ther. 2007;9:R77.
- Lee S, Moon CS, Sul D, et al. Comparison of growth factor and cytokine expression in patients with degenerated disc disease and herniated nucleus pulposus. Clin Biochem. 2009;42:1504–1511.
- Yan J, Li Q, Mao AP, et al. TRIM4 modulates type I interferon induction and cellular antiviral response by targeting RIG-I for K63-linked ubiquitination. J Mol Cell Biol. 2014;6:154–163.
- Kawai T, Akira S. Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol Med. 2011;3:513–527.
- Zhou Z, Jia X, Xue Q, et al. TRIM14 is a mitochondrial adaptor that facilitates retinoic acid-inducible gene-I-like receptor-mediated innate immune response. Proc Natl Acad Sci USA. 2014;111:E245–E254.
- Wang S, Chen Y, Li C, et al. TRIM14 inhibits hepatitis C virus infection by SPRY domain-dependent targeted degradation of the viral NS5A protein. Sci Rep. 2016;6:32336
- Nenasheva VV, Kovaleva GV, Uryvaev LV, et al. Enhanced expression of trim14 gene suppressed Sindbis virus reproduction and modulated the transcription of a large number of genes of innate immunity. Immunol Res. 2015;62:255–262.
- Su X, Wang J, Chen W, et al. Overexpression of TRIM14 promotes tongue squamous cell carcinoma aggressiveness by activating the NF-kappaB signaling pathway. Oncotarget. 2016;7:9939–9950.
- Hosono M, Shinoda Y, Hirano T, et al. Interaction of Ca(2+)-dependent activator protein for secretion 1 (CAPS1) with septin family proteins in mouse brain. Neurosci Lett. 2016;617:232–235.
- Bertrand MJ, Milutinovic S, Dickson KM, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700.
- Shedid D, Benzel EC. Cervical spondylosis anatomy: pathophysiology and biomechanics. Neurosurgery. 2007;60:S7–S13.
- Adams MA, Roughley PJ. What is intervertebral disc degeneration, and what causes it? Spine. 2006;31:2151–2161.
- Zhou GQ, Yang F, Leung VVL, et al. (v) Molecular and cellular biology of the intervertebral disc and the use of animal models. Curr Orthop. 2008;22:267–273.
- Le Maitre CL, Freemont AJ, Hoyland JA. Accelerated cellular senescence in degenerate intervertebral discs: a possible role in the pathogenesis of intervertebral disc degeneration. Arthritis Res Ther. 2007;9:R45.
- Zhao CQ, Liu D, Li H, et al. Interleukin-1beta enhances the effect of serum deprivation on rat annular cell apoptosis. Apoptosis. 2007;12:2155–2161.
- Yang D, Wang D, Shimer A, et al. Glutathione protects human nucleus pulposus cells from cell apoptosis and inhibition of matrix synthesis. Connect Tissue Res. 2014;55:132–139.
- Park JB, Park IC, Park SJ, et al. Anti-apoptotic effects of caspase inhibitors on rat intervertebral disc cells. J Bone Joint Surg Am. 2006; 88:771–779.
- Tripathi P, Aggarwal A. NF-kB transcription factor: a key player in the generation of immune response. Curr Sci. 2006;90:519–531.
- Dolcet X, Llobet D, Pallares J, et al. NF-kB in development and progression of human cancer. Virchows Arch. 2005;446:475–482.
- Chen G, Sun W, Liang Y, et al. Maternal diabetes modulates offspring cell proliferation and apoptosis during odontogenesis via the TLR4/NF-kappaB signalling pathway. Cell Prolif. 2017;50:e12324.
- Tu J, Li W, Zhang Y, et al. Simvastatin inhibits IL-1β-induced apoptosis and extracellular matrix degradation by suppressing the NF-kB and MAPK pathways in nucleus pulposus cells. Inflammation. 2017;40:725–734.
- Imamura R, Konaka K, Matsumoto N, et al. Fas ligand induces cell-autonomous NF-kappaB activation and interleukin-8 production by a mechanism distinct from that of tumor necrosis factor-alpha. J Biol Chem. 2004;279:46415–46423.
- Liu Q, Tan Q, Zheng Y, et al. Blockade of Fas signaling in breast cancer cells suppresses tumor growth and metastasis via disruption of Fas signaling-initiated cancer-related inflammation. J Biol Chem. 2014;289:11522–11535.
- Hai J, Zhu CQ, Wang T, et al. TRIM14 is a putative tumor suppressor and regulator of innate immune response in non-small cell lung cancer. Sci Rep. 2017;7:39692.
- Klumpp S, Thissen MC, Krieglstein J. Protein phosphatases types 2Calpha and 2Cbeta in apoptosis. Biochm Soc Trans. 2006;34:1370–1375.
- Zhang YQ, Herman B. ARC protects rat cardiomyocytes against oxidative stress through inhibition of caspase-2 mediated mitochondrial pathway. J Cell Biochem. 2006;99:575–588.
- Tamura S, Toriumi S, Saito J, et al. PP2C family members play key roles in regulation of cell survival and apoptosis. Cancer Sci. 2006;97:563–567.
- Klumpp S, Maurer A, Zhu Y, et al. Protein kinase CK2 phosphorylates BAD at threonine-117. Neurochem Int. 2004;45:747–752.