
Abstract
Endothelial cell (EC) dysfunction induces atherosclerotic coronary heart disease (CHD) development. Recent studies demonstrated that lncRNA NEAT1 mediates multiple biological functions of cells. How NEAT1 regulates EC function is still unclear, so this study explored the role and mechanism of NEAT1 in oxidative stress-induced ECs. The levels of NEAT1 and miR-181d-5p were measured in serum samples from ApoE−/− mice and t-BHP-treated human umbilical vein endothelial cells (HUVECs) by qRT-PCR. The potential role of NEAT1 in viability, migration and apoptosis was analyzed by CCK-8, cell metastasis, flow cytometry, dual-luciferase reporter, RNA immunoprecipitation and Western blot assays using HUVECs overexpressing NEAT1. The expression of NEAT1 was increased, but miR-181d-5p expression was decreased in serum samples from both ApoE−/− mice and t-BHP-treated HUVECs. Overexpression of NEAT1 increased viability, migration and CDKN3 expression but decreased apoptotic rates, caspase-3 activity and miR-181d-5p expression in HUVECs. In addition, NEAT1 acted as a promoter of the proangiogenic capacity of HUVECs by targeting miR-181d-5p/CDKN3. Altogether, these findings indicate that NEAT1 may exert a protective effect on HUVECs by regulating the miR-181d-5p/CDKN3A axis.
Introduction
Cardiovascular disease (CVD) is an aggressive disease with a high morbidity rate worldwide [Citation1–3]. Excessive oxidative stress and abnormal inflammatory responses accumulate endothelial cell (EC (ECs) injury [Citation4]. The initial step of CVD is EC dysfunction-induced cardiac microvascular injury [Citation5,6]. Thus, the prevention of oxidative stress-induced EC injury is needed to improve therapeutic strategies needed for improving therapeutic stratagems.
For many years, evidence has shown that lncRNAs are closely related to the pathogenesis of CVD [Citation7,8]. Wang et al. showed that long noncoding RNA AK08838 upregulates the viability of H/R cardiomyocytes via miR-30a [Citation9]. LINC00968 expression is upregulated in coronary artery disease tissues and oxLDL-treated ECs. Overexpression of LINC00968 promotes the proliferation and migration of ECs via targeting miR-9-3p [Citation10]. In uremic patients, increased expression of DKFZP434I0714 is a new potential predictor of worse CV outcomes [Citation11]. Abnormal angiogenesis is connected with the pathogenesis of CVD. The role of lncRNA in angiogenesis and EC dysfunction has also been investigated [Citation12,13]. LncRNA XIST was shown to induce oxLDL-treated EC injury by regulating the miR-320/NOD2 pathway [Citation14]. LncRNA H19 contributes to oxLDL-treated EC injury via miR-let-7/periostin. In conclusion, these results suggest that lncRNA is involved in ox-LDL-induced oxidative stress and inflammation responses in ECs. LncRNA NEAT1 is upregulated and functions as a promoter in human malignancies such as hepatocellular carcinoma [Citation15], cervical cancer [Citation16], ovarian cancer [Citation17] and others [Citation18–20]. Several studies have also demonstrated that NEAT1 is involved in EC biological behaviors. NEAT1 expression was shown to be increased in glioma endothelial cells (GECs). Knockdown of NEAT1 enhanced blood-tumor barrier permeability via miR-181d-5p [Citation21]. NEAT1 was shown to play a protective role in oxygen-glucose-deprived brain microvascular endothelial cells [Citation22]. NEAT1 also regulates immune cell functions, and NEAT1 expression was reduced in myocardial infarction patients [Citation23]. However, the biological role of NEAT1 in oxidative stress-treated HUVECs remains largely unknown. Herein, the effects of NEAT1 on the proangiogenic capacity of HUVECs were investigated by gain-of-function assays. Furthermore, the underlying mechanism of NEAT1 in t-BHP-treated HUVECs was explored.
Materials and methods
Cell culture
Primary HUVECs were maintained in endothelial growth medium (Lonza, MD, USA) with 10% FBS, 5% L-glutamine, 1% EGF and 1% heparin at 37 °C in a 5% CO2 atmosphere [Citation24]. HUVECs at passages 3–6 were used for the following experiments. Two hundred microliters of t-BHP was used to treat HUVECs for 24 h.
Cell transfection
pcDNA3.1‐NEAT1 (10 nM) miR-181d-5p mimics (20 nM), anti‐miR-181d-5p (20 nM), pcDNA3.1‐ CDKN3 (10 nM) and negative controls were provided by GenePharma (Shanghai, China). Cell transfection (1 × 105) was conducted using Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA) for 48 h, and the transfected cells were harvested for subsequent investigations.
Cell viability analysis
First, HUVECs were transfected with the designated vector and treated with t-BHP (200 μM) for 24 h. HUVEC viability was then assessed using a Cell Counting Kit (CCK)-8 assay according to the manufacturer's instructions. The absorbance (OD) at 450 nm was examined with an ELISA reader.
Cell migration and wound healing assays
First, HUVECs were transfected with the designated vector and treated with t-BHP (200 μM) for 24 h. HUVECs were then added into the upper transwell chambers (Costar, Cambridge, MA). A total of 600 µL of DMEM containing 10% FBS was added to the lower chambers. After incubation for 12 h at 37 °C, the cells were stained with 0.1% calcein M. The migrated HUVECs were calculated by a Zeiss confocal microscope.
First, HUVECs were transfected with the designated vector and treated with t-BHP (200 μM) for 24 h. HUVECs were seeded into 6-well culture plates overnight. A scratch was generated with a 200-μL pipette tip. Then, 200 μL of DMEM containing 10% FBS was added to the wells at 37 °C for 24 h. The relative gap distance was determined by a light microscope.
Cell apoptosis analysis
First, HUVECs were transfected with the designated vector and treated with t-BHP (200 μM) for 24 h. HUVEC apoptosis was analyzed by flow cytometry (Beckman Coulter, Atlanta, USA) using an Annexin V-FITC/PI apoptosis kit. HUVECs were treated with Annexin V-FITC and PI solution for 15 min following the manufacturer's instructions. HUVEC apoptosis was also measured using a caspase-3 activity kit according to the manufacturer's instructions.
RNA immunoprecipitation and biotin-tagged miR-181d-5p pulldown analysis
RNA immunoprecipitation assays were performed using an EZMagna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, Billerica, MA, USA). HUVECs were lysed with RIP lysis buffer, and cell lysates were then incubated with magnetic beads conjugated with human anti-Ago2 antibody or control mouse IgG (Abcam, Shanghai, China). Total RNA was measured by qPCR analysis.
For the biotin-tagged miR-181d-5p pull-down analysis, HUVECs were transfected with biotin-tagged miR-181d-5p for 48 h. Then, HUVEC lysates were harvested and incubated with Dynabeads™ M-280 Streptavidin at 4 °C overnight. RNA binding was measured by qPCR analysis.
Luciferase assay
HUVECs were co-transfected with NEAT1-MT or NEAT1-MUT 3’UTR and miR-181d-5p mimic or miR-NC using Lipofectamine 2000. After 48 h, the luciferase activity was assessed using a Dual-Luciferase Reporter Assay Kit (Promega, Madison, USA).
Real-time PCR analysis
Total RNA from ApoE−/−- mouse serum and t-BHP-treated HUVECs was extracted using TRIzol reagent (Invitrogen). Total RNA was transcribed using PrimeScript™ RT Master Mix (TaKaRa, Dalian, China). Then, real-time PCR experiments were performed using a SYBR® Premix Ex TaqTM II kit (TaKaRa,Dalian, China) (for NEAT1) and a TaqMan Human MiRNA assay kit (TaKaRa, Dalian, China) (for miR-181d-5p). The relative levels were determined according to the 2 − ΔΔCt method
Western blot analysis
Total protein from HUVECs was harvested by RIPA lysis buffer. Protein samples (30 µg) were separated on 10% SDS-PAGE gels, and the samples were transferred onto PVDF membranes. After blocking with 5% nonfat skim milk, the blots were incubated overnight with primary antibodies at 4 °C. The membranes were then treated with horseradish peroxidase-conjugated secondary antibody. The bands were detected using an ECL Plus Detection System (Thermo Scientific, CA, USA).
Animal experiments
ApoE−/− mouse (male, 17–22 g) were obtained from the Southern Model Animal Center (Shanghai, China). To determine the effect of NEAT1 on atherosclerosis, ApoE−/− mouse were divided randomly into a standard chow diet (control) group and a high-fat diet (HFD) group. The high-fat diet mouse were injected with control lentivirus (pcDNA3.1‐NC) or pcDNA3.1‐NEAT1 through their tail veins. The control group mouse were fed a standard chow diet, while the HFD group mice were fed a high-fat diet (1.25% cholesterol, 40% fat) for 8 weeks. Food intake and body weight were monitored. Serum samples and tissues were harvested for further investigation.
Determination of serum lipid profile
Serum was harvested by centrifugation at 3000 × g for 10 min at 4 °C and maintained at −80 °C. Serum lipid profiles were assessed according to the manufacturer's instructions.
Determination of atherosclerotic plaques
The entire aorta was excised from each mouse and fixed with 4% paraformaldehyde. The lesion areas of the aortas were detected by Oil Red O staining. The atherosclerotic plaque areas are expressed as the percent of the positive plaque area of the whole aorta as measured by Image-Pro Plus.
Statistical analysis
All data are presented as the mean ± standard deviation (SD) and analyzed with GraphPad Prism 5. Comparisons were performed with one-way ANOVA followed by Bonferroni post hoc test. Statistical significance was defined as p < .05.
Results
NEAT1 expression is increased in ApoE−/− mouse and t-BHP-treated HUVECs
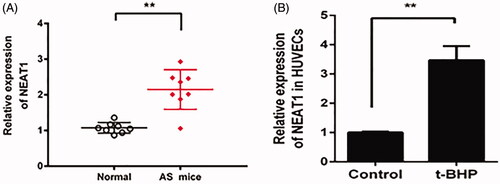
The expression pattern of NEAT1 in HFD-treated ApoE−/− mouse aortic plaques and t-BHP-treated HUVECs was measured. The quantitative PCR data showed that NEAT1 expression was increased in HFD-treated ApoE−/− mouse aortic plaques and t-BHP-treated HUVECs ()).
Figure 1. NEAT1 expression was increased in AS mouse serum, and t-BHP-treated HUVECs were analyzed by qRT-PCR. Determination of NEAT1 expression in serum samples obtained from AS mice (A) and t-BHP-treated HUVECs (B). **p < .01.
Overexpression of NEAT1 increases the proliferation and migration of t-BHP-treated HUVECs
To further elucidate how NEAT1 mediates the proangiogenic capacity of HUVECs, pcDNA3.1-NEAT1 was employed. NEAT1 expression was induced in the pcDNA3.1-NEAT1 transfection group (). The CCK-8 assay for viability demonstrated that pcDNA3.1-NEAT1 increased the viability of t-BHP-treated HUVECs (). In addition, the contribution of NEAT1 to the migration of t-BHP-treated HUVECs was analyzed by transwell and wound healing assays. As shown in , the migration ability was induced in t-BHP-treated HUVECs by NEAT1 overexpression. These data indicated that NEAT1 could promote the proangiogenic capacity of HUVECs.
Figure 2. NEAT1 promoted proangiogenic capacity while inhibiting apoptosis in t-BHP-treated HUVECs. (A) t-BHP-treated HUVECs were transfected with pcDNA3.1-NEAT1 for 48 h, and NEAT1 expression was detected by qRT-PCR. (B) HUVEC viability was evaluated by CCK-8 assay. (C) HUVEC migration was evaluated by Transwell assay. (D) HUVEC migration was evaluated by wound healing assay. (E) The apoptotic rate of HUVECs was evaluated by flow cytometry analysis. (F) Caspase-3 activity was determined in t-BHP-treated HUVECs. **p<.01.
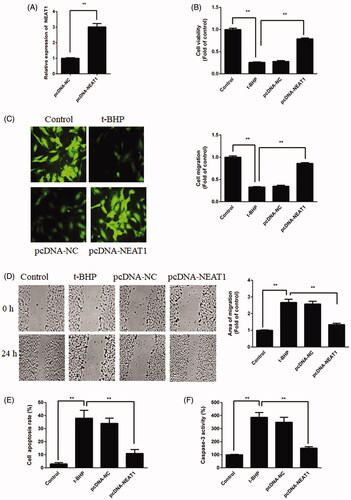
Overexpression of NEAT1 inhibits the apoptosis of t-BHP-treated HUVECs
The HUVEC apoptosis rate was analyzed by flow cytometry and caspase-3 activity. The results revealed that the apoptotic rate of t-BHP-treated HUVECs was reduced after treatment with pcDNA3.1-NEAT1 (), while t-BHP-treated HUVEC caspase-3 activity was also decreased by NEAT1 overexpression (). Altogether, these results indicated that NEAT1 suppresses t-BHP-treated HUVEC apoptosis.
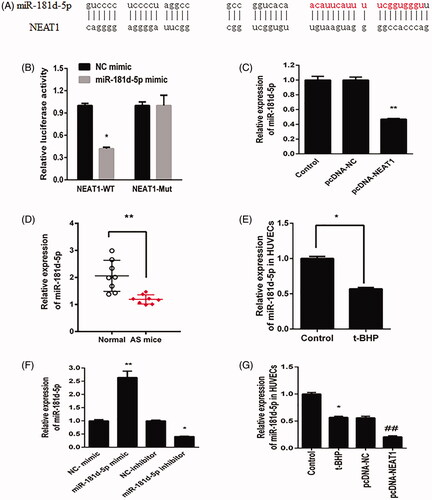
NEAT1 directly targets miR-181d-5p in HUVECs
Bioinformatics assay data revealed that NEAT1 could bind with miR-181d-5p (). To analyze in depth the relationship between NEAT1 and miR-181d-5p, a dual-luciferase reporter assay was performed. Compared with the control mimic, the miR-181d-5p mimic reduced the NEAT1-Wt plasmid luciferase activity; nonetheless, the miR-181d-5p mimic could not regulate the NEAT1-Mut luciferase activity in HUVECs (). Furthermore, HUVECs treated with pcDNA3.1-NEAT1 had inhibited miR-181d-5p expression (). In addition, both ApoE−/− mouse serum and t-BHP-treated HUVECs showed lower levels of miR-181d-5p (). The miR-181d-5p mimic increased the miR-181d-5p level, while miR-181d-5p inhibitor decreased the miR-181d-5p level in HUVECs (). Furthermore, HUVECs treated with pcDNA3.1-NEAT1 had inhibited miR-181d-5p expression in t-BHP treated HUVECs (). These results revealed that NEAT1 can directly target miR-181d-5p and negatively modulate its expression in HUVECs.
Figure 3. NEAT1 negatively regulated miR-181d-5p expression. (A) Putative binding sites between miR-181d-5p and NEAT1. (B) Relative luciferase activity in HUVECs transfected with miR-181d-5p mimic and NEAT1 wild-type (NEAT1-Wt) and mutant (NEAT1-Mut) sequences. (C) HUVECs were transfected with pcDNA3.1-NEAT1, and miR-181d-5p expression was measured by qRT-PCR. Determination of miR-181d-5p expression in samples obtained from AS mice (D) and t-BHP-treated HUVECs (E). (F) HUVECs were transfected with miR-181d-5p mimic, miR-181d-5p inhibitor and controls, and miR-181d-5p expression was measured by qRT-PCR. (G) HUVECs were transfected with pcDNA3.1-NEAT1 and control, and miR-181d-5p expression was measured by qRT-PCR. *p<.05, **p<.01, ##p<.01.
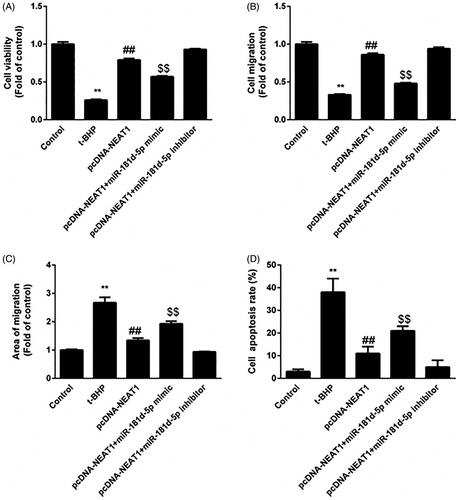
NEAT1 negatively regulates the role of miR-181d-5p in HUVECs
We further determined whether NEAT1 mediated the proangiogenic capacity of HUVECs by targeting miR-181d-5p. The effects of pcDNA3.1-NEAT1 on proliferation (), migration () and apoptosis () in t-BHP-treated HUVECs were diminished by the miR-181d-5p mimic but further enhanced by anti-miR-181d-5p. Altogether, these data revealed that NEAT1 promotes the proangiogenic capacity of t-BHP-treated HUVECs via negatively modulating miR-181d-5p.
Figure 4. miR-181d-5p inhibition of NEAT1 promotes proangiogenic capacity in t-BHP-treated HUVECs. HUVECs were transfected with pcDNA3.1-NEAT1 and miR-181d-5p mimic or anti-miR-181d-5p. (A) Cell viability was analyzed by CCK-8 assay. (B, C) HUVEC migration was evaluated by Transwell and wound healing assays. (D) The apoptotic rate of HUVECs was evaluated by flow cytometry analysis. **p < .01, ##p < .01, $$p < .01.
The NEAT1/miR-181d-5p axis regulates CDKN3 expression in HUVECs
miR-181d-5p inhibited non-small-cell lung cancer progression by targeting CDKN3A [Citation25]. Hence, we hypothesized that CDKN3 is a direct target of miR-181d-5p in HUVECs. As shown in , the miR-181d-5p mimic reduced CDKN3 expression in HUVECs, whereas anti-miR-181d-5p increased CDKN3 expression. These results demonstrated that miR-181d-5p could regulate CDKN3 expression in HUVECs.
Figure 5. NEAT1 regulated the miR-181d-5p/CDKN3A axis in t-BHP-treated HUVECs. (A) HUVECs were treated with miR-181d-5p mimic and anti-miR-181d-5p, and CDKN3 expression was determined by Western blot analysis. (B) RIP analysis for the Ago2 enrichment of NEAT1 and CDKN3 in HUVECs treated with sh-NEAT1. (C) NEAT1 and CDKN3 expression according to pull-down assay with biotin-miR-181d-5p. (D) t-BHP-treated HUVECs were transfected with pcDNA3.1-NEAT1 and miR-181d-5p mimic or anti-miR-181d-5p, and CDKN3 expression was measured by Western blot analysis. (E) HUVEC viability was examined by CCK-8 assay. (F) The migration of HUVECs was detected. (G) The apoptotic rate was determined in t-BHP-treated HUVECs. **p < .01, ##p < .01, $p < .05.
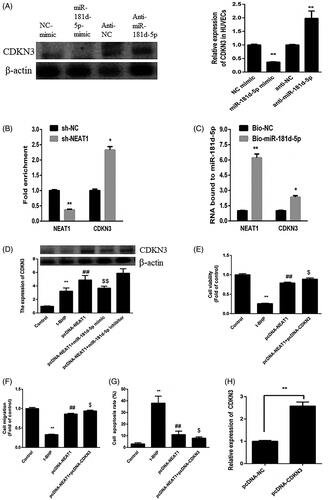
To detect whether NEAT1 directly targets the miR-181d-5p/CDKN3 signal, an RNA immunoprecipitation assay was performed. NEAT1 knockdown diminished NEAT1 enrichment but increased Ago2 enrichment of CDKN3 (). Furthermore, we verified that miR-181d-5p could bind directly to NEAT1 and CDKN3 in HUVECs and a biotinylated miR-181d-5p pull-down assay was performed. The data showed that miR-181d-5p can bind to both NEAT1 and CDKN3 in HUVECs (). Upregulation of NEAT1 increased the expression of CDKN3, which was enhanced by anti-miR-181d-5p but suppressed by the miR-181d-5p mimic ().
In addition, the effects of NEAT1 on the proangiogenic capacity of t-BHP-treated HUVECs were enhanced by increasing CDKN3 expression (). The CDKN3 expression increased after pcDNA3.1-NEAT1 transfection (). These data suggested that NEAT1 mediates the proangiogenic capacity of t-BHP-treated HUVECs by regulating miR-181d-5p/CDKN3.
NEAT1 alleviates as mouse atherosclerotic plaque formation
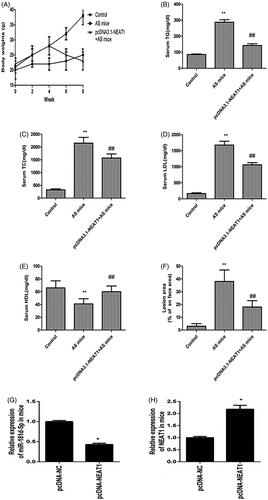
Then, we explored the protective effects of NEAT1 on atherosclerotic lesion formation in AS mouse, and there was no remarkable difference in food intake among the groups after 8 weeks (data not shown). In addition, the body weights of AS mouse were increased, whereas pcDNA3.1-NEAT1 prevented this effect (). Moreover, serum levels of TC (), TG (), and LDL () in AS mouse were increased and HDL levels were decreased (). Notably, transfection with pcDNA3.1-NEAT1 rescued all of these patterns (). AS mice had increased atherosclerotic plaque area, whereas pcDNA3.1-NEAT1 alleviated the atherosclerotic lesions in vivo (). After transfection with pcDNA3.1-NEAT1, NEAT1 levels increased, while miR-181d-5p expression was reduced in the thoracic aortic plaques of AS mouse (). These data suggested that pcDNA3.1-NEAT1 prevented atherosclerotic plaque formation in AS mouse.
Figure 6. NEAT1 suppressed atherosclerotic plaque formation in AS mice. (A) ApoE−/− mice were treated with HFD for 8 weeks and body weight was evaluated. The serum levels of TC (B), TG (C), LDL (D), and HDL (E) were detected in AS mice with NEAT1 overexpression. (F) Measurement of the atherosclerotic lesion area by Oil Red O staining. (G) miR-181d-5p expression was detected in the thoracic aortic plaques of AS mouse. (H) NEAT1 level was detected in the thoracic aortic plaques of AS mouse. *p < .05, **p < .01, ##p < .01.
Discussion
The pathogenesis of AS progresses with EC dysfunction [Citation26,27]. It is known that oxidative stress causes endothelial dysfunction [Citation28–30]. In this study, t-BHP-treated HUVECs were used to generate an AS model in vitro and the role of NEAT1 in AS progression was explored. In this study, we found that NEAT1 expression was increased in serum from AS mouse and t-BHP-treated HUVECs. Upregulation of NEAT1 induced the proangiogenic capacity but prevented apoptosis in HUVECs. An in-depth mechanism evaluation revealed that NEAT1 functions as a ceRNA to modulate the miR-181d-5p/CDKN3 axis.
Previous reports have shown that NEAT1 participates in several biological events, such as proliferation, polarization invasion and metastasis [Citation31–33]. However, the function of NEAT1 in EC dysfunction remains unknown. Our results indicated that AS mouse serum and t-BHP-treated HUVECs showed higher levels of NEAT1. To better understand the effect of NEAT1 on HUVECs, the data revealed that NEAT1 promotes the proangiogenic capacity of HUVECs. LncRNAs perform their functions via several mechanisms, such as transcriptional and post-transcriptional alterations, miRNA sponging, and, mRNA and protein regulation. NEAT1 induces colon cancer progression by binding with miR-495-3p and inducing CDK6 expression [Citation34]. NEAT1 exerts ox-LDL-induced inflammation and oxidative stress via downregulating miR-128 in macrophages [Citation35]. Knockdown of NEAT1 plays a suppressive role in cell immunity during sepsis by inducing the miR-125/MCEMP1 pathway [Citation36]. A bioinformatic analysis showed the potential binding sites between NEAT1 and miR-181d-5p. NEAT1 is located primarily in the cytoplasm. As previously reported, we also found that increased NEAT1 inhibits miR-181d-5p expression in HUVECs. Further data revealed that the miR-181d-5p mimic abrogated the effects of NEAT1 overexpression on t-BHP-treated HUVEC injury. These results demonstrated that NEAT1 might target miR-181d-5p and contribute to the angiogenesis process.
It has been indicated that miR-181d-5p is a suppressor of multiple cell processes [Citation25]. We found that AS mouse serum and t-BHP-treated HUVECs showed lower levels of miR-181d-5p. Furthermore, overexpression of NEAT1 inhibited miR-181d-5p expression. The proangiogenic role of NEAT1 in HUVECs was attenuated by a miR181d-5p mimic and further enhanced by anti-miR-181d-5p. These results suggested that NEAT1 exerts a proangiogenic role in HUVECs via negatively regulating miR-181d-5p.
Gao et al. demonstrated that miR-181d-5p inhibited non-small-cell lung cancer progression via the CDKN3/Akt pathway [Citation25]. Therefore, we demonstrated the relationship between NEAT1, miR-181d-5p and CDKN3 in HUVECs. Inhibition of NEAT1 suppressed Ago2 enrichment of NEAT1 while enhancing CDKN3 enrichment. Both NEAT1 and CDKN3 could directly bind to miR-181d-5p in HUVECs according to a pull-down assay. Moreover, NEAT1 overexpression induced CDKN3 expression, the effect of which was reversed by the miR-181d-5p mimic but further enhanced by anti-miR-181d-5p. In addition, the proangiogenic capacity of NEAT1 in HUVECs was at least partially mediated by CDKN3.
In conclusion, these findings indicated that NEAT1 expression was increased, whereas miR-181d-5p expression was decreased in AS mouse serum and t-BHP-treated HUVECs. The proangiogenic capacity of NEAT1 in HUVECs was at least partially mediated by miR-181d-5p to regulate CDKN3 expression. These findings highlight the key function of NEAT1 in AS and may provide a potential target for treating AS.
Disclosure statement
The authors did not report any conflicts of interest.
Additional information
Funding
Related Research Data
References
- Haybar H, Shahrabi S, Ghanavat M, et al. Clonal hematopoiesis: Genes and underlying mechanisms in cardiovascular disease development [Review]. J Cell Physiol. 2019;234(6):8396–8401.
- Kajikawa M, Maruhashi T, Kishimoto S, et al. Target of triglycerides as residual risk for cardiovascular events in patients with coronary artery disease- post hoc analysis of the FMD-J Study A. Circ J. 2019;83(5):1064–1071.
- Ohmura H. Triglycerides as residual risk for atherosclerotic cardiovascular disease. Circ J. 2019;83(5):969–970.
- Haybar H, Rezaeeyan H, Shahjahani M, et al. T-bet transcription factor in cardiovascular disease: attenuation or inflammation factor? [Review]. J Cell Physiol. 2019;234(6):7915–7922.
- Carracedo J, Alique M, Ramirez-Carracedo R, et al. Endothelial extracellular vesicles produced by senescent cells: pathophysiological role in the cardiovascular disease associated with all types of diabetes mellitus. Curr Vasc Pharmacol. 2018. doi:10.2174/1570161116666180820115726
- Haybar H, Shahrabi S, Rezaeeyan H, et al. Endothelial cells: from dysfunction mechanism to pharmacological effect in cardiovascular disease [review]. Cardiovasc Toxicol. 2019;19(1):13–22.
- Altesha MA, Ni T, Khan A, et al. Circular RNA in cardiovascular disease [Review]. J Cell Physiol. 2019;234(5):5588–5600.
- Wang P, Yuan Y. LncRNA-ROR alleviates hypoxia-triggered damages by downregulating miR-145 in rat cardiomyocytes H9c2 cells. J Cell Physiol. 2019. doi:10.1002/jcp.28938
- Wang JJ, Bie ZD, Sun CF. Long noncoding RNA AK088388 regulates autophagy through miR-30a to affect cardiomyocyte injury. J Cell Biochem. 2019. doi:10.1002/jcb.28300
- Wang X, Zhao Z, Zhang W, et al. Long noncoding RNA LINC00968 promotes endothelial cell proliferation and migration via regulating miR-9-3p expression. J Cell Biochem. 2018. doi:10.1002/jcb.28103
- Lai CF, Chen YT, Gu J, et al. Circulating long noncoding RNA DKFZP434I0714 predicts adverse cardiovascular outcomes in patients with end-stage renal disease. Int J Cardiol. 2019;277:212–219.
- Yu B, Wang S. Angio-LncRs: LncRNAs that regulate angiogenesis and vascular disease. Theranostics. 2018;8(13):3654–3675.
- Monteiro JP, Bennett M, Rodor J, et al. Endothelial function and dysfunction in the cardiovascular system: the long non-coding road. Cardiovasc Res. 2019. doi:10.1093/cvr/cvz154
- Xu X, Ma C, Liu C, et al. Knockdown of long noncoding RNA XIST alleviates oxidative low-density lipoprotein-mediated endothelial cells injury through modulation of miR-320/NOD2 axis. Biochem Biophys Res Commun. 2018;503(2):586–592.
- Chen S, Xia X. Long noncoding RNA NEAT1 suppresses sorafenib sensitivity of hepatocellular carcinoma cells via regulating miR-335-c-Met. J Cell Physiol. 2019. doi:10.1002/jcp.27567
- Yuan LY, Zhou M, Lv H, et al. Involvement of NEAT1/miR-133a axis in promoting cervical cancer progression via targeting SOX4. J Cell Physiol. 2019;234(10):18985–18993.
- Yong W, Yu D, Jun Z, et al. Long noncoding RNA NEAT1, regulated by LIN28B, promotes cell proliferation and migration through sponging miR-506 in high-grade serous ovarian cancer. Cell Death Dis. 2018;9(9):861.
- Li P, Huang R, Huang T, et al. Long non-coding RNA NEAT1 promotes proliferation, migration and invasion of human osteosarcoma cells. Int J Med Sci. 2018;15(11):1227–1234.
- Geng W, Guo X, Zhang L, et al. Resveratrol inhibits proliferation, migration and invasion of multiple myeloma cells via NEAT1-mediated Wnt/beta-catenin signaling pathway. Biomed Pharmacother. 2018;107:484–494.
- Zhou K, Zhang C, Yao H, et al. Knockdown of long non-coding RNA NEAT1 inhibits glioma cell migration and invasion via modulation of SOX2 targeted by miR-132 [Research Support, Non-U.S. Gov't]. Mol Cancer. 2018;17(1):105.
- Guo J, Cai H, Zheng J, et al. Long non-coding RNA NEAT1 regulates permeability of the blood-tumor barrier via miR-181d-5p-mediated expression changes in ZO-1, occludin, and claudin-5. Biochim Biophys Acta Mol Basis Dis. 2017;1863(9):2240–2254.
- Zhou ZW, Zheng LJ, Ren X, et al. LncRNA NEAT1 facilitates survival and angiogenesis in oxygen-glucose deprivation (OGD)-induced brain microvascular endothelial cells (BMECs) via targeting miR-377 and upregulating SIRT1, VEGFA, and BCL-XL. Brain Res. 2019;1707:90–98.
- Gast M, Rauch B, Haghikia A, et al. Long noncoding RNA NEAT1 modulates immune cell functions and is suppressed in early onset myocardial infarction patients. Cardiovasc Res. 2019. doi:10.1093/cvr/cvz085
- Zhang M, Xu Y, Jiang L. Irisin attenuates oxidized low-density lipoprotein impaired angiogenesis through AKT/mTOR/S6K1/Nrf2 pathway. J Cell Physiol. 2019;234(10):18951–18962.
- Gao LM, Zheng Y, Wang P, et al. Tumor-suppressive effects of microRNA-181d-5p on non-small-cell lung cancer through the CDKN3-mediated Akt signaling pathway in vivo and in vitro. Am J Physiol Lung Cell Mol Physiol. 2019;316(5):L918–L933.
- Wen J, Lin T, Wu W, et al. Tiaopi huxin recipe improved endothelial dysfunction and attenuated atherosclerosis by decreasing the expression of caveolin-1 in ApoE-deficient mice. J Cell Physiol. 2019. doi:10.1002/jcp.28184
- Zhaolin Z, Jiaojiao C, Peng W, et al. OxLDL induces vascular endothelial cell pyroptosis through miR-125a-5p/TET2 pathway. J Cell Physiol. 2019;234(5):7475–7491.
- Ito F, Sono Y, Ito T. Measurement and clinical significance of lipid peroxidation as a biomarker of oxidative stress: oxidative stress in diabetes, atherosclerosis, and chronic inflammation. Antioxidants (Basel). 2019;8(3):pii: E72.
- Li J, Zhong Z, Yuan J, et al. Resveratrol improves endothelial dysfunction and attenuates atherogenesis in apolipoprotein E-deficient mice. J Nutr Biochem. 2019;67:63–71.
- Maamoun H, Benameur T, Pintus G, et al. Crosstalk between oxidative stress and Endoplasmic Reticulum (ER) stress in endothelial dysfunction and aberrant angiogenesis associated with diabetes: a focus on the protective roles of Heme Oxygenase (HO)-1. Front Physiol. 2019;10:70.
- Tu J, Zhao Z, Xu M, et al. NEAT1 upregulates TGF-beta1 to induce hepatocellular carcinoma progression by sponging hsa-mir-139-5p. J Cell Physiol. 2018;233(11):8578–8587.
- Liu R, Tang A, Wang X, et al. Inhibition of lncRNA NEAT1 suppresses the inflammatory response in IBD by modulating the intestinal epithelial barrier and by exosome-mediated polarization of macrophages. Int J Mol Med. 2018;42(5):2903–2913.
- Zheng X, Zhang Y, Liu Y, et al. HIF-2alpha activated lncRNA NEAT1 promotes hepatocellular carcinoma cell invasion and metastasis by affecting the epithelial-mesenchymal transition. J Cell Biochem. 2018;119(4):3247–3256.
- He Z, Dang J, Song A, et al. NEAT1 promotes colon cancer progression through sponging miR-495-3p and activating CDK6 in vitro and in vivo. J Cell Physiol. 2019;234(11):19582–19591.
- Chen DD, Hui LL, Zhang XC, et al. NEAT1 contributes to ox-LDL-induced inflammation and oxidative stress in macrophages through inhibiting miR-128. J Cell Biochem. 2018. doi:10.1002/jcb.27541.
- Chen JX, Xu X, Zhang S. Silence of long noncoding RNA NEAT1 exerts suppressive effects on immunity during sepsis by promoting MicroRNA-125-Dependent MCEMP1 downregulation. IUBMB Life. 2019;71(7):956–968.