
Abstract
Ventilator has been widely used for life support, but ventilator-induced lung injury (VILI) is still a major problem. Oxidative stress has been considered as a key contributor for VILI, but the specific mechanism remains unclear. The expression of NLRP3 inflammasome in cells and inflammatory factors in the supernatant were measured. Mitochondrial ROS and TRPM2 channel currents were investigated using flow cytometry and Patch-clamp technique, respectively. TRPM2-/- and NLRP3-/- mice were used for animal experiments. Lung tissues were stained by HE and the wet-dry ratio, bronchoalveolar lavage fluid (BALF) protein, MPO (marrow peroxidase), NLRP3 inflammasome were also investigated. Knockdown of NLRP3 or Caspase-1 or treatments with SS-31 or YVAD inhibited the expression of the NLRP3 inflammasome, and reduced IL-1β and IL-18 levels in cell supernatant. These treatments suppressed the production of ROS and lowered the TRPM2 channel currents, but Rotenone exerted an opposite effect. High-tidal volume ventilation significantly increased the levels of IL-1β, IL-18, NLRP3 inflammasome, wet-dry ratio of lung, MPO and BALF protein. However, these parameters were down-regulated in TRPM2-/- and NLRP3-/- mice. These parameters were suppressed in TRPM2-/- and NLRP3-/- mice indicate that oxidative stress might promote VILI through activating NLRP3 inflammasome and TRPM2 channel.
Introduction
Mechanical ventilator has been commonly used for life support, but some complications such as acute lung injury and ventilator-induced lung injury (VILI) may be caused by it [Citation1,Citation2]. The previous report indicated that low tidal volume [Citation3] might decrease the lung injury, which implies the relationship between lung injury and mechanical ventilator, especially for high tidal volume application. Oxidative stress is believed to be closely related to ventilator-induced lung injury [Citation4], but the specific mechanism remains unclear.
Inflammation could be caused due to excessive alveolar expansion and the local collapse of the lung. The triggered inflammation cascade could lead to lung tissues injury. Inflammasomes are composed of a variety of proteins that modulate the innate immune responses [Citation5]. The main proteins include NLRP1, NLRP3, and NLRC4 [Citation6], among which NLRP3 inflammasome could be activated by noxious stimuli, such as tissue damage, metabolism, oxidative stress and infection. Meanwhile, activation of inflammasomes could lead to the production of IL-1β and IL-18, which are proven markers of inflammation induced by VILI [Citation7]. However, whether oxidative stress induces VILI through activating NLRP3 inflammasome has not been clarified.
Transient receptor potential melastatin-related 2 (TRPM2) is a non-selective cation channel with a unique C-terminal adenosine diphosphate ribose pyrophosphatase domain [Citation8,Citation9] and it is linked with Ca2+ permeability. Meanwhile, TRPM2 is a molecular sensor for reactive oxygen species (ROS) and the activation of it contribute to ROS induced brain injury [Citation10]. However, the role of the TRPM2 channel in VILI remains unclear.
Oxidative stress has been believed to be a contributor to VILI. In this study, we firstly investigated the influence of oxidative stress on NLRP3 inflammasome, TRPM2 channel, IL-1β, and IL-18. Then, the animal model of VILI was established using TRPM2-/- and NLRP3-/- mice. TRPM2-/- and NLRP3-/- mice shown significant resistance on VILI suggests that oxidative stress may influence ventilator-induced lung injury by regulating NLRP3 inflammasome and TRPM2 channel. This research may provide a new thought for the prevention of VILI by targeting NLRP3 inflammasome or TRPM2 channel.
Materials and methods
Cell culture
Alveolar macrophages (Cell Bank, Chinese Academy of Sciences, Shanghai, China) were used for in vitro study. Firstly, the dishes were pre-coated with 0.1% collagen. Then, cells (105 cells each dish) were plated on the bottom of 6-well elastic Petri dish (BioFlex, Orlando, FL, USA) in MCDB-131 medium (Caisson Labs, Logan, UT, USA) containing 10% fetal bovine serum. Cells were cultured in 37˚C with 5% CO2. Before the measurement of protein expression, cytokines concentration, ROS intensity, and TRPM2 channel current, the cells were treated firstly by SS-31 (50 nM), Rotenone (500 nM) and YVAD (5 µM) for 24 h. SS-31, Rotenone, and YVAD were purchased from Sigma (Saint Louis, MO, USA). YVAD and Rotenone were dissolved in DMSO to achieve a concentration of 1 mM and then diluted with DMEM medium to obtain the final concentrations. SS-31 was diluted using the medium to achieve final concentration.
Cell transfection
NLRP3 siRNA, Caspase-1 siRNA, and Sc siRNA were provided by GenePharma Co., Ltd (Shanghai, China). Cells were transfected with siRNA once the cells reached a confluence of 50–70%. The transfection efficiency was verified by PCR and the transfected cells were cultured for 48 h and used for subsequent experiments.
Western blotting
Cells were lysed for 30 min in RIPA buffer firstly and the total protein was measured with BCA kit (Nanjing Jiangcheng Bioengineering Institute, Nanjing, China). Proteins were denatured and separated (10 µg/lane) on 12% SDS-PAGE gel and transferred to nitrocellulose membrane (Millipore, Saint Louis, MO, USA). The membranes were incubated with primary antibodies overnight at 4 °C and then incubated in secondary antibody for 1 h at room temperature. ECL Plus detection system (Millipore, USA) was used for measuring protein expression. The gray value of immunoreactive bands was analyzed using the Quantity One software (Bio Rad, Berkeley, CA, USA). Rabbit polyclonal antibody against NLRP3 (Ab5652, 1: 1000), monoclonal antibody against ASC (Ab8400, 1: 2000) and polyclonal antibody against Caspase-1 (Ab14367, 1: 1000) were purchased from Abcam (Hong Kong, China).
Real-time PCR
Total RNA was extracted using TRIzol reagent (Cwbio, Beijing, China) and 500 ng RNA was reverse-transcribed into cDNA through Primer Script RT reagent kit (Takara Bio, Beijing, China). Real-time PCR was conducted through the SYBR Premix Ex Taq TM II kit (Takara Bio, China). The primers used for NLRP3 and Caspase-1 were listed as follows: (1) NLRP3: forward: 5′-CATCAATGCTGCTTCGACAT-3′ and reverse: 5′-TCAGTCCCACACACAGCAAT-3′; (2) Caspase-1: forward: 5′-CACAGCTCTGGAGATGGTGA-3′ and reverse: 5′-GGTCCCACATATTCCCTCCT-3′. Every sample was replicated in triplicate. The relative expression of target genes was calculated using the cycle threshold method (2-ΔΔCt). ΔΔCt = ΔCtexperiment − ΔCtcontrol, ΔCt = Cttarget gene − Ctcontrol gene. The fold change between the experimental group and the control group = 2-ΔΔCt.
Measurement of IL-1β and IL-18 using Elisa assay
The cell supernatants and serum were collected. IL-1β and IL-18 in the supernatant and serum were measured with relative ELISA kits (Nanjing Jiangcheng Bioengineering Institute, Nanjing, China).
Detection of ROS by flow cytometry
Cells were labeled in the dark with 2 mM DCFHDA–AM (Invitrogen-Molecular Probes, Carlsbad, CA, USA) for 30 min at first. After washing and re-suspending in PBS, the cells were analyzed by flow cytometer (Becton–Dickinson, Franklin Lakes, NJ, USA) at an excitation wavelength of 514 nm. Untreated cells served as controls.
TRPM2 channel current measurement
The channel currents were recorded every 5 s using EPC-10 amplifier and the data were stored in PatchMaster software (HEKA, Westfield, MA, USA) as described previously [Citation11]. Briefly, a capillary glass tube (BF150-86–10, Sutter Instruments, Novato, CA, USA) was pulled into the electrodes using a micropipette puller. The electrodes were used to contact cells under an inverted microscope using motorized micromanipulator (MP285, Sutter Instruments, USA) and sucked with a negative pressure pump to obtain GΩ seal. Immediately, after the GΩ seal was achieved, fast capacitor compensation was applied and negative pressure was applied to break the cell membrane for whole-cell recording. Then, the slow capacitor compensation was applied and the membrane capacitance and resistance were recorded. As a control, TRPM2 was activated by ADP-Ribose (0.5 mM) in the intracellular fluid after the membranes were broken. When TRPM2 channel current was stable, 2-APB was added for 5 min (or till the current was stable) and the current was recorded. Several individual cells were detected repeatedly and independently. All electrophysiological experiments were performed at room temperature.
Establishment of VILI animal model
WT C57BL/6, TRPM2-/-, and NLRP3-/- mice were purchased from Charles River (Beijing, China). All animal experimental protocols were approved by the Shandong University research ethical committee (Approval No. ECAESDUSM 2012029). All methods were performed following the relevant guidelines and regulations. Gene expression was identified using Western blot. The animals were randomly divided into three groups (normal tidal volume, high tidal volume and, control), and each group contained 6 mice. For normal tidal volume and high tidal volume groups, the animals were connected to a ventilator (Minivent, Harvard Bioscience, Holliston, MA, USA) after tracheotomy and ventilated at normal tidal volume (Vt:6 ml/kg) or high tidal volume (Vt:40 ml/kg). The inspiratory/expiratory ratio was 1:1 and respiratory frequency was adjusted to maintain PETCO2 at 35–45 mmHg. Lavage fluid and lung specimens were collected 2 h and 4 h later, respectively. The bronchoalveolar lavage fluid (BALF) was centrifuged and the supernatants were stored at −70 °C for subsequent analysis.
HE staining
The lung tissues were collected and stained as described [Citation12]. About 20% chloral hydrate (2 ml/kg) was used for the anaesthetization. Then, the right upper lung tissues were collected and stored in 4% paraformaldehyde. The sectioned tissue slices were dewaxed with xylene twice (5 min/each time), dehydrated using gradient alcohol and washed with distilled water (15 min). Sections were stained by hematoxylin for 3 min and differentiated using 2% hydrochloric acid for 10 s. After differentiation, 2% of eosin-alcohol was conducted for dyeing (3 min). A microscope could be used for observation after regular gradient alcohol dehydration and mounting.
Measurement of lung wet-dry ratio
The left lung was isolated for detecting lung wet-dry ratio. After the measurement of the wet weight of lung tissues, tissues were incubated in an oven (65 °C for 72 h). Then, the lung wet-dry ratio was calculated.
Measurement of MPO
MPO test kit (Sigma, USA) was applied to measure the MPO activity. The tissues were immersed in 2 ml buffer and homogenated by an electric homogenate machine. MPO activity was detected by Varioskan Flash multifunction plate reader (Thermo, MA, USA) at 450 nm.
Statistical analysis
Data were analyzed using SPSS software (V(0).19, IBM SPSS Statistics, Chicago, IL, USA) and expressed as means ± SD. The difference was determined using one-way or two-way ANOVA with the Tukey’s test as a post hoc test. p < 0.05 is viewed as the statistically significant difference.
Results
Oxidative stress promoted the expression of NLRP3 inflammasome and inflammatory factors
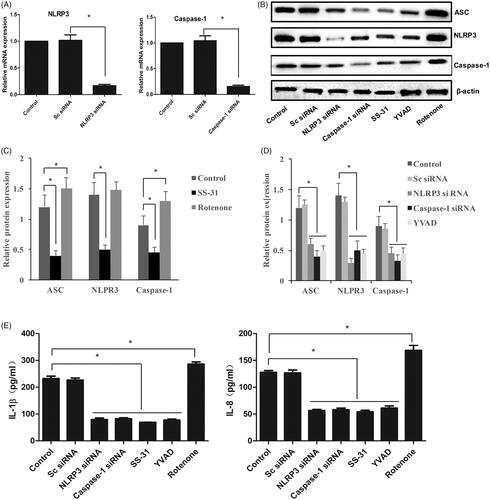
The knockdown models of NLRP3 and Caspase-1 in vitro were at first established by transfecting NLRP3-siRNA and Caspase-1-siRNA, respectively (). The components of NLRP3 inflammasome, ASC, NLRP3, and Caspase-1 were significantly suppressed by treatments with NLRP3-siRNA, Caspase-1-siRNA, or YVAD, which is an inhibitor of Caspase-1 (). Meanwhile, SS-31, an antioxidant peptide, presented a remarkable inhibitory effect on NLRP3 inflammasome (). However, the expression of ASC, NLRP3, and Caspase-1 was markedly promoted by Rotenone, which could cause oxidative stress in cells. In addition, the levels of IL-1β and IL-18 were significantly inhibited by NLRP3-siRNA, Caspase-1-siRNA, SS-31, and YVAD, but they were remarkably promoted by treatment with Rotenone (). These findings indicate that NLRP3 inflammasome and inflammatory factors could be modulated by oxidative stress.
Figure 1. Oxidative stress promoted the expression of NLRP3 inflammasome and inflammatory factors. Knockdown of NLRP3 and Caspase-1 via transfecting siRNA (A); Western blotting analysis of NLRP3 inflammasome (B); Relative protein expression of NLRP3 inflammasome after treatment with SS-31 and Rotenone (C); Relative protein expression of NLRP3 inflammasome after treatment with NLRP3 siRNA, Caspase-1 siRNA, and YVAD (D); the concentrations of IL-1β and IL-18 in cell supernatant (E). *p<.05 vs. control (Sc siRNA).
Oxidative stress led to the increase of TRPM2 channel currents and ROS production in cell
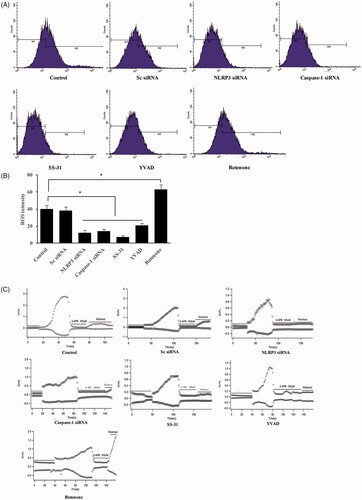
ROS has been considered to closely linked with several diseases including ventilator-induced lung injury. TRPM2 channel is a molecular sensor for ROS. We investigated the influence of oxidative stress, NLRP3-siRNA, and Caspase-1-siRNA on mitochondrial ROS and TRPM2 channel currents. We found that NLRP3-siRNA, Caspase-1-siRNA, SS-31, and YVAD significantly suppressed the production of mitochondrial ROS and TRPM2 channel currents, but Rotenone exerted an opposite effect (). Therefore, knockdown of NLRP3 or Caspase-1 might alleviate the injury caused by ROS.
Figure 2. Oxidative stress led to the increase of TRPM2 channel currents and ROS production in the cell. Measurement of ROS production with flow cytometry (A); Quantitative analysis of ROS production after different treatments (B); Measurement of TRPM2 channel current after different treatments (C). *p<.05 vs. control (Sc siRNA).
Knockdown of NLRP3 or TRPM2 downregulated the expression of NLRP3 inflammasome and inflammatory factors in vivo
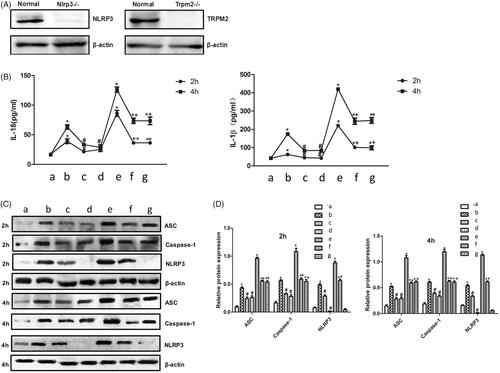
In order to investigate the regulatory role of NLRP3 andTRPM2 in ventilator-induced lung injury in vivo, we established the mice model using NLRP3-/- andTRPM2-/- mice, which was identified firstly by Western blotting (). We found that both normal and high tidal volume ventilation could remarkably increase IL-1β and IL-18 in serum and promote the expression of NLRP3 inflammasome in lung tissue () compared with group control. However, NLRP3-/- andTRPM2-/- mice presented significant resistance on either normal or high tidal volume ventilation. The contents of IL-1β and IL-18 in serum and NLRP3 inflammasome in lung tissue were markedly suppressed in group NLRP3-/- andTRPM2-/- mice compared with group WT C57BL/6 mice ().
Figure 3. Knockdown of NLRP3 or TRPM2 downregulated the expression of NLRP3 inflammasome and inflammatory factors in vivo. Identification of TRPM2-/- and NLRP3-/- mice with Western blotting method (A); Measurement of IL-1β and IL-18 in serum after normal or high tidal volume mechanical ventilator (B); Measurement of NLRP3 inflammasome protein expression in lung tissue after normal or high tidal volume mechanical ventilator (C); Quantitative analysis of NLRP3 inflammasome protein expression in lung tissue after normal or high tidal volume mechanical ventilator (D). a: WT C57BL/6 mice treated with sham operation; b: WT C57BL/6 mice treated with normal tidal volume ventilation; c: TRPM2-/- mice treated with normal tidal volume ventilation; d: NLRP3-/- mice treated with normal tidal volume ventilation; e: WT C57BL/6 mice treated with high tidal volume ventilation; f: TRPM2-/- mice treated with high tidal volume ventilation; g: NLRP3-/- mice treated with high tidal volume ventilation. * p<.05 vs. control; #p<.05 vs. normal tidal volume ventilation; + p<.05 vs. high-tidal volume ventilation.
Knockdown of NLRP3 or TRPM2 alleviated the ventilator-induced lung injury
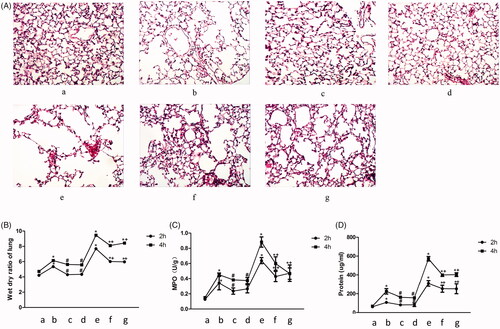
Meanwhile, histological changes of lung tissue, wet-dry ratio of the lung, MPO, and BALF protein were investigated. Distention of alveoli and inflammatory cell infiltration was observed in group WT C57BL/6 mice treated by ventilation, especially the treatment with high tidal volume ventilation. However, a significant decrease of structural disruption of the lung tissue was observed in the NLRP3-/- andTRPM2-/- mice. Meanwhile, VILI-induced edema and infiltration of inflammatory cells were decreased remarkably in the NLRP3-/- andTRPM2-/- mice (). Wet-dry ratio has been widely used to detect pulmonary edema. MPO and BALF protein are also two important indicators of lung injury. Wet-dry ratio of the lung, MPO, and BALF protein were remarkably increased in WT C57BL/6 mice after normal or high tidal volume ventilation, but less lung injury was observed in NLRP3-/- andTRPM2-/- mice. These findings indicate that NLRP3-/- andTRPM2-/- mice present significant resistance to ventilator-induced lung injury.
Figure 4. Knockdown of NLRP3 or TRPM2 alleviated the ventilator-induced lung injury. Histological changes of lung tissue stained by HE, infiltration of inflammatory cells were marked with black arrows (A); Measurement of the wet-dry ratio of lung in VILI models (B); Measurement of MPO in VILI models (C); Measurement of BALF protein in VILI models (D). a: WT C57BL/6 mice treated with sham operation; b: WT C57BL/6 mice treated with normal tidal volume ventilation; c: TRPM2-/- mice treated with normal tidal volume ventilation; d: NLRP3-/- mice treated with normal tidal volume ventilation; e: WT C57BL/6 mice treated with high tidal volume ventilation; f: TRPM2-/- mice treated with high tidal volume ventilation; g: NLRP3-/- mice treated with high tidal volume ventilation. *p<.05 vs. control; #p<.05 vs. normal tidal volume ventilation; +p<.05 vs. high tidal volume ventilation.
Discussion
Mechanical ventilation is increasingly used in life support and routine anesthesia operations [Citation13], but the VILI has always been a thorny problem. Studies have shown that the experimental mice suffered fatal emphysema on the high airway pressure condition [Citation14]. Meanwhile, using the normal airway pressure, ventilator still can cause a significant increase of permeability in alveolar capillary and even lung injury [Citation15]. Unfortunately, the specific mechanism of VILI has not been clearly clarified.
Our study suggests that knockdown of NLRP3 and Caspase-1 could inhibit the expression of ASC, NLRP3, and Caspase-1 (). Similar results were observed after treatments with SS31 (Anti-oxidant) and YVAD (Caspase-1 inhibitor). Down-regulation of NLRP3 and Caspase-1 could result in the decreased formation of NLRP3 inflammasome. Rotenone is the inhibitor of mitochondrial complex I electron transfer chain and possesses the potential oxidative toxicity. Rotenone remarkably up-regulated the expression of ASC, NLRP3, and Caspase-1 (). The previous study indicated that mitochondrial damage could increase ROS production and further activate NLRP3 inflammasome [Citation16]. Therefore, the activation of inflammasome might be caused by Rotenone-induced increase of ROS in the mitochondrion.
Studies have shown that ROS is an important activator for NLRP3 inflammasome [Citation17] and ROS inhibitors or scavengers could inhibit the expression of NLRP3 inflammasome. We also found the increase of ROS was significantly suppressed by treatment with NLRP3 siRNA, Caspase-1 siRNA, SS31, or YVAD. Mitochondrial damage and ROS production could aggravate lung injury and the inhibition of mitochondrial ROS production may be a potential prevention target for VILI.
The transient receptor potential (TRP) channels are important cation channels on the cell membrane, which are permeable to calcium ions. The TRP channels have 6 subfamilies: TRPC, TRPY, TRPM, TRPA, TRPP and TRPML [Citation18,Citation19]. TRPM2 channel is a class of non-selective cation channel of calcium ions, which is highly expressed in the brain tissues [Citation20]. Studies have shown that stimulation of adverse factors such as inflammation, ischemia, and hypoxia could suppress TRPM2 channel, and further enhance the survival of astrocytes [Citation21]. Meanwhile, knockdown of TRPM2 switched cells from cell death to autophagy for survival in response to oxidative stress [Citation22]. In our study, TRPM2 channel currently declined markedly after treatment with NLRP3 siRNA and Caspase-1 siRNA, suggesting that NLRP3 and Caspase-1 play an important role in regulating the TRPM2 channel.
IL-1β and IL-18 are two interleukin-1 family cytokines, which are the main regulators of the host immune response [Citation23]. It was reported that IL-1β and IL-18 precursors could be cleaved by Caspase-1 to generate mature IL-1β and IL-18, and then the inflammatory reaction was triggered as a protection mechanism [Citation24,Citation25]. We found that the release of IL-1 β and IL-18 was inhibited after treatment with NLRP3 siRNA, Caspase-1 siRNA, SS31, or YVAD. However, Rotenone markedly increased the levels of IL-1β and IL-18, which indicates that antioxidation and knockdown of NLRP3 and Caspase-1 may decrease the release of IL-1β and IL-18. These results were also verified in animal models.
The animal study indicated that high tidal volume ventilation resulted in the remarkable increase of ASC, NLRP3, Caspase-1, MPO, wet-dry ratio, and BALF protein, but NLRP3-/- and TRPM2-/- mice shown resistance on high tidal volume ventilation. These findings indicate that NLRP3 and TRPM2 are closely involved in VILI.
In summary, this research demonstrates that knockdown of NLRP3 or treatment with antioxidant significantly could significantly decrease ROS production in the mitochondrion and TRPM2 channel currents. Meanwhile, NLRP3-/- and TRPM2-/- mice presented remarkable resistance on the VILI induced by mechanical ventilation. Therefore, inhibition of NLRP3 or TRPM2 may be a potential strategy for the prevention of VILI.
Author contributions
XA and JW conceived and designed the experiments; XS and XY performed the experiments; DL, YH, and HC analyzed the data; XA and JW wrote the paper. All authors read and approved the final manuscript.
| Abbreviations | ||
| VILI | = | Ventilator-induced lung injury |
| BALF | = | bronchoalveolar lavage fluid |
| MPO | = | Bone marrow peroxidase |
| TRPM2 | = | Transient receptor potential M2 |
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- Cressoni M, Gotti M, Chiurazzi C, et al. Mechanical power and development of ventilator-induced lung injury. Anesthesiology. 2016;124:1100–1108.
- Cabrera-Benitez NE, Laffey JG, Parotto M, et al. Mechanical ventilation-associated lung fibrosis in acute respiratory distress syndrome: a significant contributor to poor outcome. Anesthesiology. 2014;121:189–198.
- Zhang Y, Gao J, Wang CJ, et al. Low tidal volume ventilation preconditioning ameliorates lipopolysaccharide-induced acute lung injury in rats. Acta Anaesthesiol Scand. 2016;60:780–789.
- Wagner J, Strosing KM, Spassov SG, et al. Sevoflurane posttreatment prevents oxidative and inflammatory injury in ventilator-induced lung injury. PLOS One. 2018;13:e0192896.
- Son S, Hwang I, Han SH, et al. Advanced glycation end products impair NLRP3 inflammasome-mediated innate immune responses in macrophages. J Biol Chem. 2017;292:20437–20448.
- Guo Q, Wu Y, Hou Y, et al. Cytokine secretion and pyroptosis of thyroid follicular cells mediated by enhanced NLRP3, NLRP1, NLRC4, and AIM2 inflammasomes are associated with autoimmune thyroiditis. Front Immunol. 2018;9:1197.
- Aung NY, Ohe R, Meng H, et al. Specific neuropilins expression in alveolar macrophages among tissue-specific macrophages. PLOS One. 2016;11:e0147358.
- Perraud AL, Schmitz C, Scharenberg AM. TRPM2 Ca2+ permeable cation channels: from gene to biological function. Cell Calcium. 2003;33:519–531.
- Inamura K, Sano Y, Mochizuki S, et al. Response to ADP-ribose by activation of TRPM2 in the CRI-G1 insulinoma cell line. J Membr Biol. 2003;191:201–207.
- Li X, Yang W, Jiang LH. Alteration in intracellular Zn(2+) homeostasis as a result of TRPM2 channel activation contributes to ROS-induced hippocampal neuronal death. Front Mol Neurosci. 2017;10:414.
- Chen GL, Zeng B, Eastmond S, et al. Pharmacological comparison of novel synthetic fenamate analogues with econazole and 2-APB on the inhibition of TRPM2 channels. Br J Pharmacol. 2012;167:1232–1243.
- Yang X, Sun X, Chen H, et al. The protective effect of dopamine on ventilator-induced lung injury via the inhibition of NLRP3 inflammasome. Int Immunopharmacol. 2017;45:68–73.
- Bignami E, Di Lull A, Saglietti F, et al. Routine practice in mechanical ventilation in cardiac surgery in Italy. J Thorac Dis. 2019;11:1571–1579.
- Kuchnicka K, Maciejewski D, Ventilator A, Lung I. Uszkodzenie płuc związane z wentylacją mechaniczną. Anaesthesiol Intensive Ther. . 2013;45:164–170.
- Plotz FB, Slutsky AS, van Vught AJ, et al. Ventilator-induced lung injury and multiple system organ failure: a critical review of facts and hypotheses. Intensive Care Med. 2004;30:1865–1872.
- Kepp O, Galluzzi L, Kroemer G. Mitochondrial control of the NLRP3 inflammasome. Nat Immunol. 2011;12:199–200.
- Minutoli L, Puzzolo D, Rinaldi M, et al. ROS-mediated NLRP3 inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxid Med Cell Longev. 2016;2016:1.
- Montell C. TRP channels in Drosophila photoreceptor cells. J Physiol (Lond). 2005;567:45–51.
- Lopez-Romero AE, Hernandez-Araiza I, Torres-Quiroz F, et al. TRP ion channels: proteins with conformational flexibility. Channels (Austin). 2019;13:207–226.
- Yamamoto S, Shimizu S, Kiyonaka S, et al. TRPM2-mediated Ca2 + influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med. 2008;14:738–747.
- Chen Y, Swanson RA. Astrocytes and brain injury. J Cereb Blood Flow Metab. 2003;23:137–149.
- Wang Q, Huang L, Yue J. Oxidative stress activates the TRPM2-Ca(2+)-CaMKII-ROS signaling loop to induce cell death in cancer cells. Biochim Biophys Acta Mol Cell Res. 2017;1864:957–967.
- van de Veerdonk FL, Netea MG, Dinarello CA, et al. Inflammasome activation and IL-1beta and IL-18 processing during infection. Trends Immunol. 2011;32:110–116.
- Schneider KS, Groß CJ, Dreier RF, et al. The inflammasome drives GSDMD-independent secondary pyroptosis and IL-1 release in the absence of caspase-1 protease activity. Cell Rep. 2017;21:3846–3859.
- Dungan LS, Mills KH. Caspase-1-processed IL-1 family cytokines play a vital role in driving innate IL-17. Cytokine. 2011;56:126–132.