
Abstract
Cardiovascular disease is recognized as a leading cause of death worldwide, but the risk of death is 2–3 times higher for individuals with diabetes. NLRP3 inflammasome activation is a leading pathway of vascular damage, and new treatment methods are needed to reduce NLRP3 inflammasome expression, along with a detailed understanding of how those treatments work. In a series of assays on human vascular endothelial cells that were exposed to high concentrations of free fatty acids (FFA) to induce a diabetes-like environment, we found a significant impact of cilostazol, a vasodilator widely used to treat blood flow problems and well-tolerated medication. To our knowledge, this study is the first to demonstrate the effects of cilostazol in primary human aortic endothelial cells. We found that cilostazol significantly reduced NLRP3 inflammasome activation, as well as the activity of other related and harmful factors, including oxidative stress, expression of NADPH oxidase 4 (NOX-4), thioredoxin-interacting protein (TxNIP), high mobility group box 1 (HMGB-1), interleukin 1β (IL-1β) and IL-18. Cilostazol also protected the functionality of sirtuin 1 (SIRT1), which serves to restrict NLRP3 inflammasome activity, when exposure to FFAs would have otherwise impaired its function. Thus, it appears that cilostazol’s mechanism of action in reducing NLRP3 inflammasome activation is an indirect one; it protects SIRT1, which then allows SIRT1 to perform its regulatory job. Cilostazol has potential as an already-available, well-tolerated preventive medication that may alleviate some of the adverse vascular effects of living with diabetes. The findings of the present study lay the groundwork for further research on the potential of cilostazol as a safe and effective treatment against diabetic endothelial dysfunction and vacular disease.
Introduction
Cardiovascular disease is recognized as a leading cause of death worldwide, but the risk of death by cardiovascular disease is 2–3 times higher for individuals with diabetes [Citation1]. Thus, it is urgent to identify ways by which the cardiovascular sequelae of diabetes can be prevented. Interventions targeting glucose levels, blood pressure, and oxidative stress do not reduce the incidence of cardiovascular deaths among diabetics [Citation2]. However, activation of the nucleotide oligomerization domain (NOD)-like receptor pyrin domain-containing 3 (NLRP3) inflammasome is known to be one of the leading pathways through which the diabetic cardiovascular system is compromised, and there exists direct evidence that inhibiting NLRP3 inflammasome activation reduces inflammation, making it a ripe target for intervention [Citation2–4]. NLRP3 inflammasome inhibitors exist but have been shown to be largely ineffective in clinical settings [Citation5]. Type I interferons (IFNs) have good efficacy but come with substantial side effects in nearly every organ system [Citation6]. In contrast, the phosphodiesterase-3 inhibitor cilostazol, a vasodilator widely used to treat blood flow problems, has anti-inflammatory effects in vascular smooth muscle cells and its side effect profile is mild [Citation7,Citation8]. Cilostazol protects against vascular dysfunction in diabetes, but its mechanisms are not well understood [Citation9]. The purpose of this study to explore the impact of cilostazol on NLRP3 inflammasome activation, and the mechanisms by which that impact occurs.
Plasma levels of free fatty acids (FFA) are considerably increased in type II diabetes patients and are considered to be a highly accurate diagnostic indicator of disease progression [Citation10]. High FFA levels are a key indicator of cardiovascular risk and have been shown to cause NLRP3 inflammasome activation [Citation11,Citation12]. In the present study, we administered high FFAs to induce NLRP3 inflammasome activation and examined the impact of cilostazol on several variables including oxidative stress, production of proinflammatory cytokines, and the involvement of the class III histone deacetylase sirtuin 1 (SIRT1). Oxidative stress is well recognized as an inducer of chronic inflammation and diabetic endothelial dysfunction [Citation2]. NADPH oxidase 4 (NOX-4) is an enzyme that promotes the production of reactive oxygen species (ROS), thereby bringing on substantial oxidative stress and enhancing endothelial dysfunction [Citation13]. Previous research has shown that suppression of NOX-4 reduces oxidative stress-induced vascular degeneration [Citation14]. Thioredoxin-interacting protein (TxNIP) is a key part of the mechanism by which NLRP3 is activated [Citation15]. It is a known mediator of NLRP3 inflammasome-induced damage to endothelial cells and promotes oxidative stress by inhibiting the antioxidant thioredoxin [Citation16,Citation17]. High mobility group box 1 (HMGB1) protein can be characterized as an alarmin that signals the need for initiation of the inflammatory response, thereby contributing to chronic inflammation and endothelial dysfunction [Citation18]. Interleukins 1-beta (IL-1β) and 18 (IL-18) are proinflammatory cytokines [Citation19] produced by NLRP3 that play a critical role in the pathogenesis of numerous inflammatory diseases. The NLRP3 inflammasome manufactures pro-IL-1β and pro-IL-18, which become mature IL-1β and IL-18 upon cleavage induced by caspase 1 [Citation3,Citation20]. SIRT1, a known protector against vascular harm [Citation21], is typically inhibited by NLRP3 and rescue of SIRT1 expression is regarded as a promising strategy to mitigate NLRP3 inflammasome-induced endothelial dysfunction. Our findings demonstrate that cilostazol significantly ameliorates oxidative stress and inflammation by ameliorating activation of the NLRP3 inflammasome via rescue of SIRT1 function. Thus, cilostazol may serve as a safe and effective therapy against endothelial dysfunction and vascular disease induced by exposure to high FFA.
Materials and methods
Cell culture and treatment
Human aortic endothelial cells (HAECs) were obtained from Lonza (Basel, Switzerland). HAECs were cultured in endothelial growth media (EGM2) in low passage numbers (<10) in a 5% CO2 humidified incubator at 37 °C and stimulated with high FFA (1 mM) in the presence or absence of 5 or 10 µM cilostazol.
MitoSOX staining
HAECs were exposed to 1 mM FFAs in the presence or absence of 5 or 10 µM cilostazol for 36 h. After being washed 3 times with PBS, cells were incubated with 5 μM MitoSOX Red for 1 h at 37 °C in darkness. Fluorescence signals were visualized using a fluorescence microscope.
Immunocytochemistry of 4-HNE
Intracellular levels of 4-HNE were evaluated based on immunofluorescence. Briefly, HAECs were pre-incubated with high FFAs (1 mM) for 36 h in the presence or absence of 5 or 10 µM cilostazol. HAECs were then fixed with 4% paraformaldehyde for 10 min at RT followed by permeabilization with 0.1% Triton X-100 in Tris-buffered saline and Tween 20 (TBST) for 15 min. Cells were then blocked with 5% bovine serum albumin (BSA) and 2.5% FBS in TBST, followed by incubation with the primary anti-4HNE monoclonal antibody overnight at 4 °C. After being washed 3 times, cells were incubated with Alexa-488 conjugated secondary antibodies for 1 h at room temperature (RT).
Real-time polymerase chain reaction (PCR)
Total RNA was extracted from HAECs using a micro RNeasy Micro Kit in accordance with the manufacturer’s instructions (Qiagen (Cat.74004), USA). RNA concentrations were quantified using Nanodrop. A total of 1 μg RNA was used to synthesize complementary DNA (cDNA) using iScript™ Reverse Transcription Supermix (BioRad, #1708840) for quantitative real-time PCR analysis (Invitrogen). SYBR-based real-time PCR experiments were performed to detect the total transcripts of mRNA using the ABI 7500 platform. The experimental results were analyzed by normalizing to GAPDH using the 2−△△Ct method
Western blot analysis
HAECs under the different treatment conditions were lysed using radioimmunoprecipitation assay (RIPA) buffer with protease and phosphatase inhibitors. Nuclear extracts were obtained by removing the cytoplasm via cell lysis with hypotonic buffer. A total of 20 μg cell lysates or nuclear extracts were immobilized via 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The separated protein mixtures were then transferred to polyvinylidene fluoride (PVDF) membranes and blotted against their specific antibodies as well as their corresponding secondary antibodies. Immunoblots were visualized using Pierce™ ECL Plus western blotting substrate (Catalog # 32132). The primary antibodies used in this study are:
Enzyme-linked immunosorbent assay (ELISA)
The secreted levels of IL-1β, IL-18, and HMGB-1 were measured by collecting the culture media of the HAECs for analysis. ELISA kits were purchased from R&D Systems and the procedure was performed in accordance with the manufacturer’s instructions. 96-plate reader spectrometry was used to collect the data.
Statistical analysis
Data are expressed as means ± standard derivation (SD). One-way analysis of variance (ANOVA) followed by Bonferroni post-test comparisons was used to determine statistical significance. p ≤ .05 was considered statistically significant.
Results
Cilostazol ameliorates FFA-induced oxidative stress
Oxidative stress induced by chronic exposure to FFA is recognized as a contributing factor in cardiovascular diseases, including diabetes-associated vascular disease [Citation22]. In the present study, we began by determining the effects of cilostazol treatment on the levels of three key indicators of oxidative stress—production of ROS, 4-HNE, and NOX4—in HAECs exposed to high FFA for 36 h to mimic chronic high FFA exposure. As shown by the results of MitoSOX Red staining in , high FFA exposure increased the level of mitochondrial ROS to roughly 3.3-fold, which was reduced by treatment with 5 and 10 µM cilostazol to only 2.1-fold and 1.4-fold, respectively. Similarly, the results of immunohistochemistry in demonstrate that high FFA increased the level of 4-HNE to 3.7-fold, which was reduced by the two doses of cilostazol in a dose-dependent manner to 2.2-fold and 1.5-fold, respectively. Next, we employed real-time PCR and western blot analysis to determine the effects of cilostazol on high FFA-induced expression of NOX4 at the mRNA and protein levels, respectively. As shown in , exposure to high FFA for 36 h resulted in a 4.1-fold increase in mRNA expression of NOX4, which was reduced to only 2.5- and 1.8-fold, respectively. At the protein level, high FFA exposure increased NOX4 expression of 3.7-fold, which was reduced by cilostazol in a dose-dependent manner to only 2.2- and 1.5-fold. These findings demonstrate a potent antioxidant capacity of cilostazol against high FFA-induced oxidative stress in endothelial cells.
Figure 1. Cilostazol inhibits high free fatty acids (FFA)-induced oxidative stress in human aortic endothelial cells (HAECs). Cells were stimulated with high FFAs (1 mM) with or without cilostazol (5, 10 μM) in HAECs for 36 h. (A). Mitochondrial ROS as examined by MitoSOX staining; (B). 4-Hydroxynonenal (4-HNE) as examined by immunocytochemistry (*, #, $, p < .01 vs. previous group).
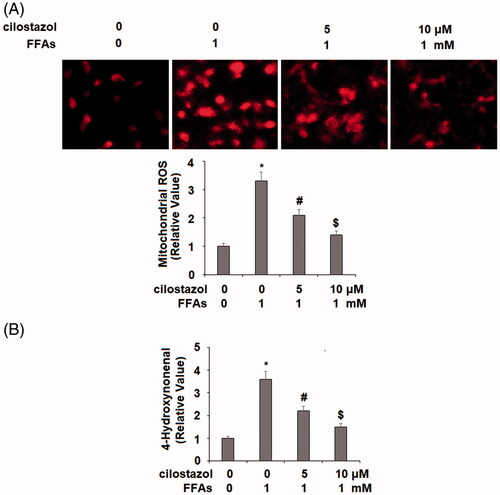
Figure 2. Cilostazol reduces FFA-induced expression of NADPH oxidase 4 (NOX4) in HAECs. Cells were stimulated with high FFAs (1 mM) with or without cilostazol (5, 10 μM) in HAECs for 36 h. (A). mRNA of NOX4 as examined by real-time PCR analysis; (B). Protein expression of NOX4 as examined by western blot analysis (*, #, $, p < .01 vs. previous group).
Cilostazol reduces FFA-induced activation of the NLRP3 inflammasome
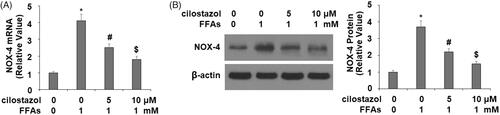
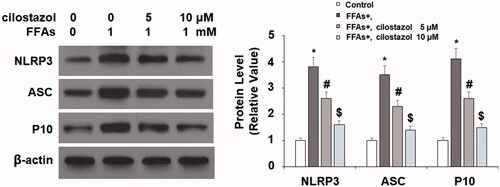
Next, we investigated the effects of cilostazol on high FFA-induced activation of the NLRP3 inflammasome by measuring the levels of TxNIP and HMGB1. As demonstrated by the results of real-time PCR and western blot analysis in , exposure to high FFA increased the expression of TxNIP to 3.8- and 3.5-fold at the mRNA and protein levels, respectively. Meanwhile, the introduction of 5 and 10 µM cilostazol potently ameliorated this effect in a dose-dependent manner, reducing the mRNA expression of TxNIP to only 2.7- and 1.9-fold, and the protein expression of TxNIP to 2.5- and 1.3-fold, respectively. The results in demonstrate a similar effect of cilostazol on high FFA-induced expression of HMGB1. Exposure to high FFA for 36 h increased the secretion of HMGB1 to 4.2-fold baseline, which was ameliorated by treatment with the two doses of cilostazol to only 2.5- and 1.4-fold, thereby demonstrating strong inhibition of HMGB1 expression. Finally, we employed western blot analysis to determine the effects of cilostazol on high FFA-induced activation of the NLRP3 inflammasome by measuring the protein expressions of NLRP3, ASC, and cleaved caspase 1 (P10). As shown in , exposure to high FFA increased NLRP3 expression to 3.8-fold, expression of ASC to 3.5-fold, and P10 expression to 4.1-fold. Meanwhile, the two doses of cilostazol significantly reduced the expression of these three factors in a dose-dependent manner. NLRP3 expression was reduced to 2.6- and 1.6-fold, ASC expression to 2.3- and 1.4-fold, and P10 expression to 2.6- and 1.5-fold.
Figure 3. Cilostazol reduces FFA-induced expression of TxNIP in HAECs. Cells were stimulated with high FFAs (1 mM) with or without cilostazol (5, 10 μM) in HAECs for 36 h. (A). mRNA level of TxNIP as examined by real-time PCR; (B). Protein level of TxNIP as examined by western blot analysis (*, #, $, p < .01 vs. previous group).
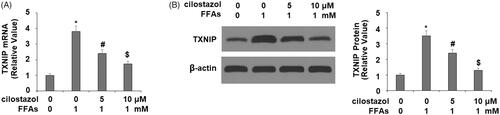
Figure 4. Cilostazol reduces FFA-induced release of high mobility group box-1 protein (HMGB-1) in HAECs. Cells were stimulated high FFAs (1 mM) with or without cilostazol (5, 10 μM) in HAECs for 36 h. Secretion of HMGB1 (1, 4.2, 2.5 1.4) as measured by ELISA (*, #, $, p < .01 vs. previous group).
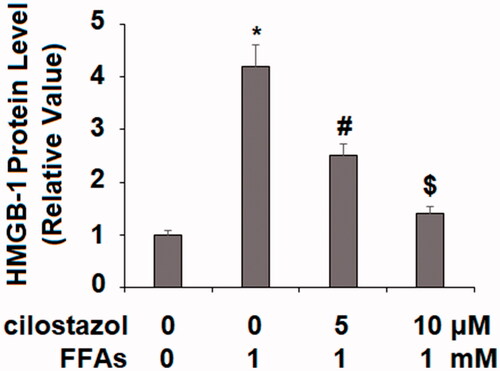
Figure 5. Cilostazol prevents FFA-induced activation of NLRP3 inflammasome in HAECs. Cells were stimulated with high FFAs (1 mM) with or without cilostazol (5, 10 μM) in HAECs for 36 h. Expression of NLRP3, ASC, and cleaved caspase 1 (P10) was measured by western blot analysis (*, #, $, p < .01 vs. previous group).
Cilostazol reduces the production of IL-1β and IL-18
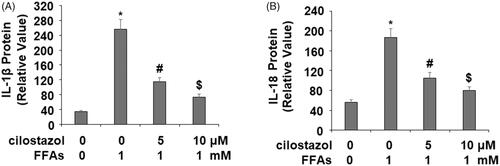
Next, we confirmed that inhibition of NLRP3 inflammasome activation by cilostazol suppressed the production of IL-1β and IL-18. As shown by the results of ELISA in , the baseline concentration of IL-1β was 34.3 pg/ml, which was increased to 256.6 pg/ml upon exposure to high FFA for 36 h. However, treatment with the two doses of cilostazol exerted a strong inhibitory effect, reducing the concentration of IL-1β to only 115.4 and 73.9 pg/ml in a dose-dependent manner. Similarly, the concentration of IL-18 was 56.2 pg/ml at baseline which was increased to 186.7 pg/ml by exposure to high FFA for 36 h, while the two doses of cilostazol significantly reduced the concentration of IL-18 to 105.4 and 79.6 pg/ml, respectively. Thus, our findings confirm that inhibition of NLRP3 inflammasome activation by cilostazol exerts a potent anti-inflammatory effect.
Figure 6. Cilostazol inhibits FFA-induced secretion of IL-1β and IL-18 in HAECs. Cells were stimulated high FFAs (1 mM) with or without cilostazol (5, 10 μM) in HAECs for 36 h. protein secretion of (A) IL-1β and (B) IL-18 was measured by ELISA (*, #, $, p < .01 vs. previous group).
The effects of cilostazol are mediated through SIRT1
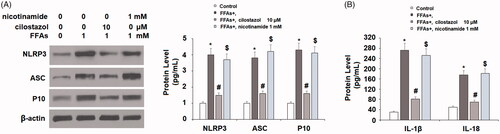
Next, we investigated the effects of cilostazol on high FFA-induced reduced expression of the NLRP3 inhibitor SIRT1. As shown in , the results of real-time PCR show that exposure to high FFA reduced the expression of SIRT1 to roughly 45%, which was recovered to 71% and 92% baseline at the mRNA level. Concordantly, the results of western blot analysis show that the protein expression of SIRT1 was reduced to 42% by high FFA exposure, while the two doses of cilostazol rescued SIRT1 protein expression to roughly 69% and 88% in a dose-dependent manner. Finally, we performed a SIRT1 inhibition experiment using nicotinamide to determine whether the effects of cilostazol are dependent on SIRT1. Briefly, HAECs were stimulated with 1 mM high FFA in the presence or absence of 10 μM cilostazol or 1 mM nicotinamide for 36 h. As demonstrated by the results of western blot analysis in , the protein expression of NLRP3 was increased to 4-fold by high FFA exposure, which was reduced to 1.5-fold by the addition of cilostazol, while inhibition of SIRT1 by nicotinamide abolished the effects of cilostazol, resulting in 3.7-fold expression of NLRP3. Meanwhile, ASC and P10 expression were respectively increased to 3.8- and 4.3-fold by high FFA alone, decreased to 1.6-fold for both factors in the presence of high FFA plus cilostazol, and increased to 4.2- and 4.1-fold in the presence of high FFA with cilostazol plus nicotinamide. Concordantly, the results of ELISA in demonstrate that the inhibition of SIRT1 by nicotinamide exerted a similar effect on the protein secretion of IL-1β and IL-18. These findings demonstrate that expression of SIRT1 plays a vital role in facilitating the effects of cilostazol against high FFA-induced activation of the NLRP3 inflammasome.
Figure 7. Cilostazol protected against FFA-induced reduction of SIRT1 in HAECs. Cells were stimulated with high FFAs (1 mM) with or without cilostazol (5, 10 μM) in HAECs for 36 h. (A). mRNA of SIRT1 as determined by real-time PCR analysis; (B). Protein of SIRT1 as determined by western blot analysis (*, #, $, p < .01 vs. previous group).
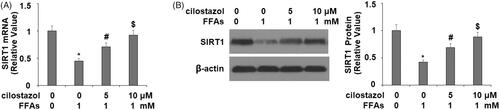
Figure 8. Inhibition of SIRT1 by nicotinamide abolished the inhibitory effects of cilostazol in FFA-induced activation of NLRP3 inflammasome. HAECs were stimulated high FFAs (1 mM) in the presence or absence of cilostazol (10 μM) or nicotinamide (1 mM) for 36 h. (A). Protein expression of NLRP3, ASC, and cleaved caspase 1 (P10) as determined by western blot analysis; (B) Protein secretion of IL-1β and IL-18 as measured by ELISA (*, #, $, p < .01 vs. previous group).
Discussion
In this study, using primary human endothelial cells exposed to high FFA, we examined the impact of cilostazol on several variables related to cardiovascular risk, including oxidative stress, expression of proinflammatory cytokines, activation of the NLRP3 inflammasome, and the involvement of SIRT1. Our findings demonstrate a powerful effect of cilostazol at clinically relevant doses to inhibit all of these aspects of endothelial dysfunction. Oxidative stress is a major contributing factor in a wide range of diseases. In diabetes-related cardiovascular disease, increased levels of mitochondrial ROS and the reactive lipid 4-HNE have been shown to promote chronic inflammation and fibrosis in various tissues and drive the disease progression of diabetes [Citation23,Citation24]. Recent studies have demonstrated the ability of cilostazol to mitigate overproduction of ROS and 4-HNE in hepatocytes and oocytes [Citation25,Citation26]. Concomitant treatment with ginkgo balboa extract and cilostazol has been shown to potentially reduce vascular disease by suppressing the development of atherosclerotic lesions and oxidative stress in ApoE mice fed a high-fat diet [Citation27]. Concordantly, our findings demonstrate that treatment with cilostazol alone significantly inhibits overproduction of ROS and 4-HNE induced by prolonged exposure to high FFA, thereby providing further evidence of a strong antioxidant effect of cilostazol. Additionally, we demonstrate that the antioxidant capacity of cilostazol may be a result of its suppression of high FFA-induced overexpression of NOX4, a key promoter of ROS production.
Increased expression of TxNIP and HMGB1 is recognized as a major event in high FFS-induced NLRP3 inflammasome activation. TxNIP also plays a key role in oxidative stress by inhibiting the expression of the antioxidants Trx-1 and Trx-2, thereby exacerbating mitochondrial ROS production. Expression of TxNIP has been shown to be involved in the regulation of numerous processes, including the inflammatory response, cellular redox balance, lipid metabolism, and cellular apoptosis [Citation27]. However, to our knowledge, this is the first study to investigate the effects of cilostazol on TxNIP expression. Our findings demonstrate a strong ability of cilostazol to inhibit the expression of TxNIP, thereby further downregulating oxidative stress induced by high FFA exposure. HMGB1 is another key component of NLRP3 inflammasome activation. Recent research has demonstrated that HMGB1 has two separate redox states—a chemotactic reduced form and a proinflammatory disulfide form, the latter of which is involved in activating the NLRP3 inflammasome [Citation28]. A recent study showed that cilostazol could suppress the secretion of HMGB1 induced by lipopolysaccharide exposure through AMPK activation and p38 MAPK expression [Citation29]. Our results also demonstrate the ability of cilostazol to downregulate HMGB1 expression induced by high FFA.
Preventing activation of the NLRP3 inflammasome is considered a valuable treatment approach against numerous chronic inflammatory diseases, including while numerous studies have suggested inhibition of NLRP3 inflammasome activation as a means to mediate the inflammatory response, a safe and reliable treatment has yet to be pinpointed. Cilostazol was originally prescribed as an antiplatelet agent and to ameliorate intermittent claudication. Recently, it has also been shown to prevent the development of foot ulcers in type II diabetes patients with peripheral vascular disease [Citation30]. Contemporary research using a rat model suggests that cilostazol can improve age-related endothelial dysfunction via suppression of oxidative stress, increased bioavailability of nitric oxide, and EDHF-like vasorelaxation. Cilostazol treatment has also been shown to be effective in preventing the development of atherosclerosis by downregulating the expression of cytokines and chemokines induced by glucose [Citation31]. In the present study, we demonstrate for the first time to our knowledge, that cilostazol can inhibit activation of the proinflammatory NLRP3 inflammasome induced by high FFA exposure, thereby significantly suppressing endothelial dysfunction due to chronic inflammation and oxidative stress. Importantly, we also show that SIRT1 plays a vital role in this beneficial effect of cilostazol, as evidenced by the results of our SIRT1 knockdown experiment using nicotinamide. Rescue of SIRT1 is considered an attractive treatment option in numerous diseases, and cilostazol has been shown to increase SIRT1 activation in neuronal cells, osteoclasts, hepatocytes, and others [Citation32–34]. However, our study is the first to our knowledge to demonstrate that cilostazol can increase SIRT1 expression in primary HAECs, thereby opening the way to a new potential use for cilostazol in SIRT1-mediated amelioration of vascular diseases. Further reseach is required to better understand the mechanisms through which cilostazol exerts these remarkable antioxidant, anti-NLRP3, and pro-SIRT1 effects in the context of high FFA-induced vascular disease.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Barquera S, Pedroza-Tobías A, Medina C, et al. Global overview of the epidemiology of atherosclerotic cardiovascular disease. Arch Med Res. 2015;46(5):328–338.
- Sharma A, Tate M, Mathew G, et al. Oxidative stress and NLRP3-inflammasome activity as significant drivers of diabetic cardiovascular complications: therapeutic implications. Front Physiol. 2018;9:114.
- Lebreton F, Berishvili E, Parnaud G, et al. NLRP3 inflammasome is expressed and regulated in human islets. Cell Death Dis. 2018;9(7):726
- Abderrazak A, El Hadri K, Bosc E, et al. Inhibition of the Inflammasome NLRP3 by arglabin attenuates inflammation, protects pancreatic β-cells from apoptosis, and prevents type 2 diabetes mellitus development in ApoE2Ki mice on a chronic high-fat diet. J Pharmacol Exp Ther. 2016;357(3):487–494.
- Shao B-Z, Xu Z-Q, Han B-Z, et al. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015;6:262.
- Sleijfer S, Bannink M, Van Gool AR, et al. Side effects of interferon-alpha therapy. Pharm World Sci. 2005;27(6):423–431.
- Weintraub WS. The vascular effects of cilostazol. Can J Cardiol. 2006;22:56B–60B.
- Aoki C, Hattori Y, Tomizawa A, et al. Anti-inflammatory role of cilostazol in vascular smooth muscle cells in vitro and in vivo. J Atheroscler Thromb. 2010;17(5):503–509.
- Ahn C, Lee H, Park S, et al. Decrease in carotid intima media thickness after 1 year of cilostazol treatment in patients with type 2 diabetes mellitus. Diabetes Res Clin Pract. 2001;52(1):45–53.
- Spiller S, Blüher M, Hoffmann R. Plasma levels of free fatty acids correlate with type 2 diabetes mellitus. Diabetes Obes Metab. 2018;20(11):2661–2669.
- Legrand-Poels S, Esser N, L’Homme L, et al. Free fatty acids as modulators of the NLRP3 Inflammasome in obesity/type 2 diabetes. Biochem Pharmacol. 2014;92(1):131–141.
- Qi Y, Du X, Yao X, et al. Vildagliptin inhibits high free fatty acid (FFA)-induced NLRP3 inflammasome activation in endothelial cells. Artif Cells Nanomed Biotechnol. 2019;47(1):1067–1074.
- Kuroda J, Ago T, Matsushima S, et al. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci USA. 2010;107(35):15565–15570.
- Wang L, Li X, Zhang Y, et al. Oxymatrine ameliorates diabetes-induced aortic endothelial dysfunction via the regulation of eNOS and NOX4. J Cell Biochem. 2018;120:7323–7332
- Mohamed IN, Hafez SS, Fairaq A, et al. Thioredoxin-interacting protein is required for endothelial NLRP3 inflammasome activation and cell death in a rat model of high-fat diet. Diabetologia. 2014;57(2):413–423.
- Liu Y, Lian K, Zhang L, et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res Cardiol. 2014;109(5):415.
- Shah A, Xia L, Goldberg H, et al. Thioredoxin-interacting protein mediates high glucose-induced reactive oxygen species generation by mitochondria and the NADPH oxidase, Nox4, in mesangial cells. J Biol Chem. 2013;288(10):6835–6848.
- Wang Y, Zhong J, Zhang X, et al. The role of HMGB1 in the pathogenesis of type 2 diabetes. J Diabetes Res. 2016;2016:1–11.
- Dinarello CA. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am J Clin Nutr. 2006;83(2):447S–455S
- Van de Veerdonk FL, Netea MG, Dinarello CA, et al. Inflammasome activation and IL-1β and IL-18 processing during infection. Trends Immunol. 2011;32(3):110–116.
- Ota H, Akishita M, Eto M, et al. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J Mol Cell Cardiol. 2007;43(5):571–579.
- Cai L, Kang YJ. Oxidative stress and diabetic cardiomyopathy. Cardiovasc Toxicol. 2001;1(3):181–193.
- Kayama Y, Raaz U, Jagger A, et al. Diabetic cardiovascular disease induced by oxidative stress. IJMS. 2015;16(10):25234–25263.
- Xiao M, Zhong H, Xia L, et al. Pathophysiology of mitochondrial lipid oxidation: role of 4-hydroxynonenal (4-HNE) and other bioactive lipids in mitochondria. Free Radic Biol Med. 2017;111:316–327.
- Xie X, Xu X, Sun C, et al. Protective effects of cilostazol on ethanol-induced damage in primary cultured hepatocytes. Cell Stress Chaperones. 2018;23(2):203–211.
- Mihalas BP, De Iuliis GN, Redgrove KA, et al. The lipid peroxidation product 4-hydroxynonenal contributes to oxidative stress-mediated deterioration of the ageing oocyte. Sci Rep. 2017;7(1):6247.
- Ding C, Zhao Y, Shi X, et al. New insights into salvianolic acid A action: regulation of the TXNIP/NLRP3 and TXNIP/ChREBP pathways ameliorates HFD-induced NAFLD in rats. Sci Rep. (1) 2016;6:28734.
- Frank MG, Weber MD, Fonken LK, et al. The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming: a role for the NLRP3 inflammasome. Brain Behav Immun. 2016;55:215–224.
- Chang KC. Cilostazol inhibits HMGB1 release in LPS-activated RAW 264.7 cells and increases the survival of septic mice. Thromb Res. 2015;136(2):456–464.
- de Franciscis S, Gallelli L, Battaglia L, et al. Cilostazol prevents foot ulcers in diabetic patients with peripheral vascular disease. Int Wound J. 2015;12(3):250–253.
- Moreira HS, Lima-Leal GA, Santos-Rocha J, et al. Phosphodiesterase-3 inhibitor cilostazol reverses endothelial dysfunction with ageing in rat mesenteric resistance arteries. Eur J Pharmacol. 2018;822:59–68.
- Lee HR, Shin HK, Park SY, et al. Cilostazol upregulates autophagy via SIRT1 activation: reducing amyloid-β peptide and APP-CTFβ levels in neuronal cells. PloS One. 2015;10(8):e0134486.
- Park SY, Lee SW, Kim HY, et al. Suppression of RANKL-induced osteoclast differentiation by cilostazol via SIRT1-induced RANK inhibition. Biochim Biophys Acta. 2015;1852(10):2137–2144.
- Kabil SL. Beneficial effects of cilostazol on liver injury induced by common bile duct ligation in rats: role of SIRT 1 signaling pathway. Clin Exp Pharmacol Physiol. 2018;45(12):1341–1350.