
Abstract
MicroRNAs (miRNAs) play a key role in various pathological processes like atrial fibrillation (AF), which is a common cardiac arrhythmia. Exosomes are essential information carrier in the intercellular communication. Therefore, this study aimed to investigate the effects of exosomal miR-320d on cardiomyocytes with AF and related mechanisms. To do this, AMSCs were transfected with miR-320d mimics, AMSCs-derived exosomes were co-cultured with cardiomyocytes with AF. MTT, TUNEL staining, flow cytometry, real-time PCR, western blots, and luciferase reporter assays were performed. The results revealed that miR-320d expression was decreased in AF cardiomyocytes. AF increased apoptosis and reduced cell viability in cardiomyocytes. By transfection with miR-320d mimics, the miR-320d level was increased in AMSCs, exosomes and cardiomyocytes, which reversed the effect of AF on cardiomyocytes. STAT3 was down-regulated in AF cardiomyocytes and was a direct target gene of miR-320d. Inhibition of STAT3 abolished the effect of modified exosomes in cardiomyocytes, causing decreased apoptosis and increased cell viability. Taken together, the results suggested that exosomal miR-320d was associated with AF cardiomyocytes apoptosis and cell viability and that the effect of miR-320d on cardiomyocytes is STAT3-dependent. Therefore, this study provides a novel understanding of the molecular basis of AF and provides insight into therapeutic strategies for AF.
Introduction
Atrial fibrillation (AF) is the most common cardiac arrhythmia, affecting about 1% of young patients and up to 9% of patients at least 75 years old [Citation1]. In patients with or without cardiovascular conditions, AF is induced by triggers in the pulmonary vein [Citation2]. Reports suggest that AF is associated with increased cardiovascular morbidity and mortality [Citation3]. From the aetiological point of view, AF is a manifestation of complex aberrancies in multiple levels, including molecular, cellular, electrical and structural alterations [Citation4]. Furthermore, various risk factors may cause AF, such as heart failure, hypertension, and mitral valve disease [Citation5].
Growing evidence reveals that AF is related to structural and electrical remodelling that are both critical for the initiation and maintenance of AF [Citation6,Citation7]. Moreover, apoptosis, a type of programmed cell death, is a complex process of cell self-destruction, characterizing abnormal cell shape, membrane blebbing and chromatin degradation [Citation8]. In AF studies, atrial HL-1 cardiomyocytes with tachypacing showed enhanced caspase activity which suggests increased apoptosis [Citation9]. In congestive heart failure-induced rapid atrial pacing models, atrial cells were observed to have a high level of apoptosis [Citation10,Citation11], implying the close correlation between the high ventricular rate and cell death in AF.
Exosomes are specialized endocytic-origin vesicles in nano-size [Citation12] and play essential roles in intercellular communication via DNA, RNAs and proteins [Citation13]. To date, exosomes have been widely applied as the carrier to deliver functional microRNAs (miRNAs) to the recipient cells in vitro [Citation14,Citation15]. Adipose tissue-derived mesenchymal stem cells (AMSCs) are regarded as an ideal source to generate exosomes [Citation16]. Thus, AMSCs can be a promising mediator to deliver miRNAs in exosome-mediated intercellular communication [Citation16]. For example, miR-181-5P transfected-AMSCs engineered exosomes to inhibit hepatic fibrosis in hepatic stellate cells [Citation17]. Also, exosomes generated from miR-122-treated AMSCs sensitize the hepatocellular carcinoma cells when given a chemotherapy treatment [Citation18]. Due to its high efficacy, it may be worthwhile to apply AMSCs-exosome approach to AF patients.
In addition to the relationship between miRNAs and exosomes, miRNAs, 21–24 nucleotides, are a class of endogenous and small non-coding RNAs that exert essential roles in the regulation of gene expression [Citation19]. As an important post-transcriptional regulator, miRNAs can bind 3′-UTR of target genes, thereby either repressing subsequent translation or inducing mRNA degradation [Citation20]. It is becoming more evident that miRNAs play significant roles in the regulatory mechanisms to modulate various biological processes [Citation19]. In AF patients, miR-26 significantly reduces AF, leading to increased K+ current which is a hallmark of atrial electrical remodelling [Citation21]. The muscle-specific miRNA, miR-133, has been reported to regulate cardiac ion channels via down-regulating the ether-a-go-go-related gene protein (ERG) level and K+ current [Citation22]. Also, miR-320d, a member of the miR-320 family, is associated with cell growth and apoptosis, for example, in glioma cells, overexpression of miR-320d inhibits cell growth while enhances apoptosis [Citation23]. Furthermore, forced expression of miR-320d increases staurosporine and tumour necrosis factor α-induced apoptosis in smooth muscle cells [Citation24]. However, the effect of miR-320d on cardiomyocytes is still unclear. Therefore, the objectives of this study were to investigate the function of miR-320d in cardiomyocytes of AF mice and whether AMSCs-exosome is an effective way to deliver miR-320d to cardiomyocytes.
Materials and methods
Animal
In this study, experimental protocols were approved by the Ethical Review Board of Cangzhou Central Hospital. All animal procedures and care were performed according to the Animal Care and Treatment Administration of the National Ministry of Health and the Ethics Committee in Cangzhou Central Hospital. Healthy male C57BL/6 mice (24.2 ± 2.1 g, 8 weeks) were purchased from the Animal Center Laboratory at Peking University (Beijing, China). All mice were individually housed at 20–26 °C with 40–70% relative humidity and a 12:12-h light–dark cycle (6:00 am to 6:00 pm). Water and food were given ad libitum.
AF induction
Wild-type mice were anesthetized via intraperitoneal injection of sodium pentobarbital (dose: 40 mg/kg) and supported by artificial respiration. Body temperature was kept at 37 °C and monitored for the entire procedure. The AF induction protocol has been previously described [Citation25]. The criteria of successful AF induction were defined as rapid irregular atrial rhythm lasting more than one second.
Cardiomyocytes isolation
Primary neonatal mice myocytes were isolated from newborn heart ventricles by trypsin solution. Briefly, hearts of newborn C57BL/6 mice were isolated aseptically after decapitation. The hearts were then cultured in serum-free Dulbecco’s modified Eagle’s medium (DMEM; Nissui Pharmaceutical, Tokyo, Japan) and were digested by 0.25% trypsin solution. The cell suspension was centrifuged at 2500 rpm for 3 min and resuspended in DMEM containing 10% foetal bovine serum (FBS; Gibco BRL, Grand Island, NY, USA). The non-adherent cardiomyocytes were plated into non-coated culture flasks (Thermo Fisher Scientific, Waltham, MA, USA). Cardiomyocytes were cultured under a condition of 5% CO2 at 37 °C.
AMSCs isolation
Subcutaneous adipose tissue was obtained from six male C57BL/6 mice. Subcutaneous adipose tissue was collected as previously described [Citation26] and was placed in MesenPRO® RS™ Medium (1% antibiotic-antimycotic; Gibco BRL, Grand Island, NY, USA). Next, AMSCs (passages 3–6) were identified by western blots of AMSC surface markers: PE-labelled cluster designation 34 (CD34), CD44, CD45, CD73, CD90, CD105 and CD166 (BD Biosciences Pharmingen, San Diego, CA, USA) [Citation27]. To test the differentiation potential (Gibco BRL, Grand Island, NY, USA), adipogenesis treatment was administered to AMSCs. The differentiated adipocytes were stained with Oil Red O as previously described [Citation28].
Plasmids and AMSC transfection
For transfection, AMSCs were cultured in the DMEM containing 10% FBS and 1% antibiotic-antimycotic (Gibco BRL, Grand Island, NY, USA). Bovine exosomes and protein aggregates were removed by ultracentrifugation at 100,000 g and 4 °C for 16 h. Next, AMSCs (1 × 106) were seeded in the DMEM overnight. Then, AMSCs were transfected with plasmids of miR-320d mimic (sense: 5′-AAAAGCUGGGUUGAGAGGA-3′) or control mimics (sense: UUCUCCGAACGUGUCACGUTT) by using Lipofectamine™ 2000 kit in accordance with the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA).
AMSC exosomes isolation
After 48-h transfection, AMSCs-derived exosomes were isolated from the AMSCs supernatant using the ExoQuick-TC Kit (System Biosciences, Mountain View, CA, USA) in accordance with the manufacturer’s instructions. The AMSCs-derived exosomes were assessed by western blots of exosome surface markers: CD9, CD63 and CD105 (BD Biosciences Pharmingen, San Diego, CA, USA) [Citation29]. Precipitates from AMSCs DMEM were used as the negative control. Exosome pellets were resuspended in PBS solution at a 5 μg/μL protein concentration.
Quantitative real-time PCR
Total RNAs were isolated using Trizol Reagent (Aidlab, Beijing, China). The cDNA was synthesized using the RNA PCR Core Kit (Applied Biosystems, Foster City, CA, USA) in accordance with the manufacturer’s instructions. Expression of miR-320d was measured using the TaqMan MicroRNA Assay (Life Technologies, Grand Island, NY, USA). The primers were used as followed: GAPDH (forward, 5′-TATGATGATATCAAGAGGGTAGT-3′; reverse, 5′-TGTATCCAAACTCATTGTCATAC-3′) and miR-320d (forward, 5′-AAAAGCTGGGTTGAGAGGA-3′; reverse, 5′-TCCTCTCAACCCAGCTTTT-3′). Real-time PCR reactions were performed on ABI7900/illumina (Applied Biosystems by Life Technologies, Grand Island, NY, USA). Real-time PCR data were analyzed using the ΔΔCT method [Citation30], and GAPDH was used as the reference gene.
Cardiomyocytes transfection
The miR-320d mimics and negative control were obtained from RiboBio (Guangzhou, China). The signal transducer and activator of transcription-3 (STAT3)-containing pCDNA3.1 and STAT3-siRNA (25 nmol/L) were obtained from Invitrogen (Invitrogen, Shanghai, China). The transfection procedure was performed using the Lipofectamine™ 2000 Transfection Reagent (Invitrogen, Waltham, MA, USA) following the manufacturer’s instructions. The transfection efficacy was evaluated by real-time PCR or western blots.
MTT assay
Cardiomyocytes with AF were plated in 6-well plates and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Abcam, Cambridge, UK) was performed to assess cardiomyocytes viability according to the manufacturer’s instructions. The absorbance was measured by an Infinite 200 PRO (Tecan, Morrisville, NC, USA) plate reader at 490 nm.
TUNEL staining
Cardiomyocytes were collected and fixed in 4% paraformaldehyde in PBS with 0.12 mM sucrose for 20 min. The apoptosis level was assessed by a TUNEL apoptosis detection kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. Apoptosis was visualized via fluorescence microscopy (AmScope, Irvine, CA, USA).
Flow cytometry
Cardiomyocyte apoptosis was measured by using an Annexin Vnexin Vn was measured bas mea (Thermo Fisher Scientific, Waltham, MA, USA) in accordance with the manufacturers’ instructions. Annexin V-FITC/propidium iodide (PI) and FITC positive cardiomyocytes were evaluated by a flow cytometry assay (BD Biosciences, San Diego, CA, USA).
Luciferase reporter assay
The target gene of miR-320d was predicted by the online tool, TargetScan (www.targetscan.org). For this study, STAT3 was predicted to be a potential target gene of miR-320d. The luciferase reporter assay (Promega, Fitchburg, WI, USA) was performed to identify if STAT3 3′-UTR sequence contained the miR-320d binding site. The protocol was performed in accordance with the manufacturers’ instructions.
Western blot
Total proteins were isolated using cell lysis buffer (Beyotime Institute of Biotechnology, Shanghai, China). Protein extracts were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE; 8–12%) and then were transferred to polyvinylidene difluoride (PVDF) membranes. Next, PVDF membranes were blocked with 5% skimmed milk in PBS (0.05%) Tween 20 for 2 h at room temperature. The primary antibodies of STAT3, caspase-3, Bax, Bcl-2 and GAPDH were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Quantification was assessed by the Uvitec Alliance software (Eppendorf, Hamburg, Germany).
Statistical analysis
Data were presented as mean ± standard error. Statistical analysis was performed using SPSS v.18.0 software (SPSS Inc., Chicago, IL, USA). Differences between groups were analyzed with Student’s t-test. Difference was considered to be significant at p < .05. The sample size in each treatment group was 3 for the entire study.
Results
Identification of AMSCs and AMSCs-derived exosomes
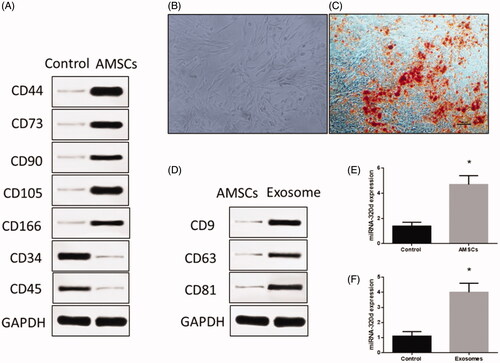
In this study, AMSCs were isolated from subcutaneous adipose tissue. In general, CD molecules were used as cell surface markers to identify the AMSCs. The results revealed that CD44, CD73, CD90, CD105 and CD166 were up-regulated while CD34 and CD45 were down-regulated in AMSCs (), suggesting that the AMSCs were correctly isolated from mice adipose tissues. Then, AMSCs were given adipogenic induction treatment for 14 days. According to the images from Oil Red O positive staining, AMSCs could differentiate to adipose tissue (), which suggests AMSCs have normal differentiation potential. Furthermore, surface markers of exosomes were detected and the results revealed that the expressions of CD9, CD63 and CD81 significantly increased in AMSCs-derived exosomes (), suggesting these exosomes could be used for subsequent experiments.
Figure 1. Isolation of adipose tissue-derived mesenchymal stem cells (AMSCs) and exosomes. (A) The AMSCs surface markers detected by western blots. (B) Representative image of AMSCs. (C) Oil Red O staining in cardiomyocyte administered the adipogenesis treatment. Scale bar = 50 μm. (D) The exosome surface markers detected by western blots. (E) miRNA-320d expression in AMSCs. (F) miRNA-320d expression in exosomes. Values are means ± SEM. For each experiment, sample size n ≥ 3. (*) denotes difference from control group (p < .05).
MiR-320d-modified AMSCs and exosomes
After cell surface marker identification, AMSCs were transfected with miR-320d mimics for 48 h. The miR-320d expression was evaluated using real-time PCR in modified AMSCs and exosomes. The results suggested that miR-320d expression significantly increased in AMSCs () and exosomes (), implying that miR-320d mimics successfully elevated miR-320 expression in AMSCs-derived exosomes.
Down-regulation of miR-320d promoted apoptosis in cardiomyocytes with AF
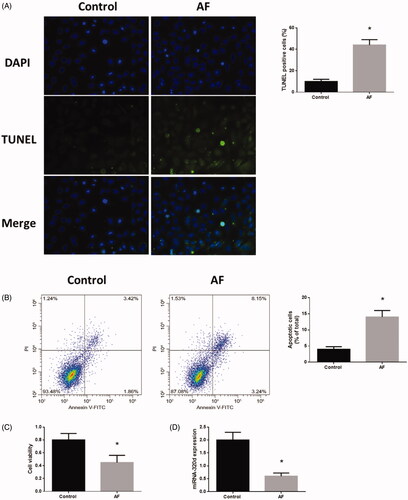
Annexin V-FITC/PI and TUNEL staining assays revealed that cardiomyocytes with AF displayed elevated levels of apoptosis (). Using the MTT assay, reduced cell viability was found in cardiomyocytes (). Meanwhile, miR-321d expression was decreased in cardiomyocytes with AF compared to control cells ().
Figure 2. Down-regulation of miR-320d was associated with the regulation of apoptosis and cell viability in cardiomyocytes with AF. (A) Apoptosis level measured by TUNEL staining assays. (B) Apoptosis level measured by Annexin V-FITC/propidium iodide (PI) assays. (C) Cell viability measured by MTT assays. (D) miRNA-320d expression in cardiomyocytes with AF. Values are means ± SEM. For each experiment, sample size n ≥ 3. (*) denotes difference from control group (p < .05).
Apoptosis reduced in cardiomyocytes with AF co-cultured with modified exosomes
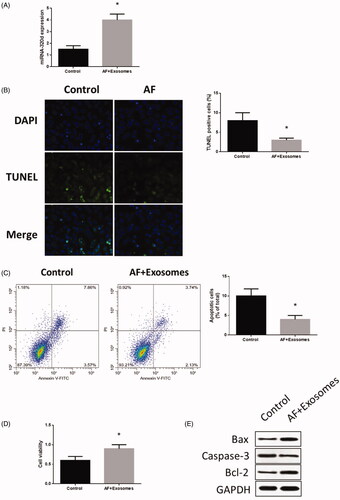
When cardiomyocytes with AF was administrated to miR-320d mimics-modified exosomes, the expression of miR-320d was increased (), leading to dampened apoptosis () and increased cell viability (). These results suggest that the effect of AF on cardiomyocytes was reversed from increased expression of miR-320d induced by engineered exosomes. To further explore the mechanisms underlying miR-320d-related apoptosis, western blots were performed to detect apoptosis-associated proteins. The results revealed that expression of Bax and caspase-3 increased while the level of Bcl-2 reduced in cardiomyocytes with AF (), suggesting that the Bax/Bcl-2/caspase-3 pathway may mediate AF-induced apoptosis in cardiomyocytes.
Figure 3. MiR-320d increased in cardiomyocytes co-cultured with modified exosomes. (A) miRNA-320d expression in co-cultured modified exosomes. (B) Apoptosis level measured by TUNEL staining assays. (C) Apoptosis level measured by Annexin V-FITC/propidium iodide (PI) assays. (D) Cell viability measured by MTT assays. (E) Apoptotic protein expressions detected by western blots. Values are means ± SEM. For each experiment, sample size n ≥ 3. (*) denotes difference from control group (p < .05).
MiR-320d promoted apoptosis in cardiomyocytes by targeting STAT3
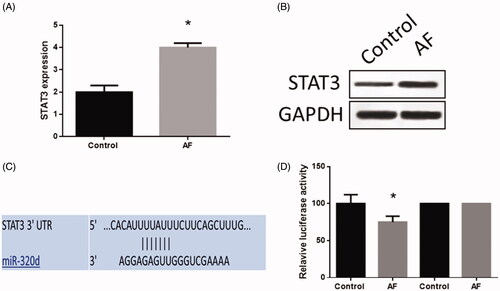
In cardiomyocytes, the expression of STAT3 was increased (). Through the online analysis tool, TargetScan (www.targetscan.org), a binding site of miR-320d was found in the 3′-UTR of STAT3 (). The luciferase reporter assay was performed to determine the mechanic relationship between STAT3 and miR-320d. The results indicated that the luciferase activity was decreased in cardiomyocytes with the wild-type 3′-UTR sequence while there was no change of luciferase activity in cells with the mutant 3′-UTR sequence (), which suggested STAT3 was a target gene of miR-320d in cardiomyocytes with AF.
Figure 4. MiR-320d targeted STAT3 in cardiomyocytes with AF. (A) STAT3 mRNA expression detected by real-time PCR. (B) STAT3 protein expression detected by western blots. (C) The predictive binding sequence of miR-320d in the 3′-UTR of STAT3. (D) Relative luciferase activity. Values are means ± SEM. For each experiment, sample size n ≥ 3. (*) denotes difference from control group (p < .05).
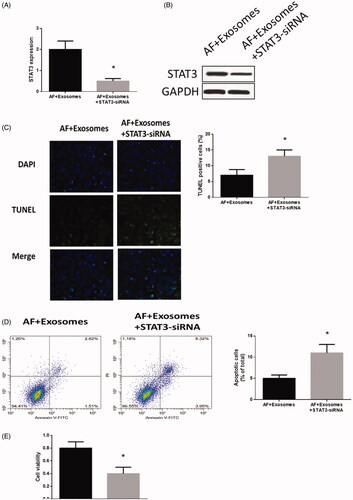
Down-regulation of STAT3 inhibited apoptosis in cardiomyocytes with AF
To further investigate the function of STAT3 in cardiomyocytes with AF, STAT3-siRNA was applied to cardiomyocytes treated with miR-320d mimics-modified exosomes. The expression of STAT3 was significantly decreased (). In addition, the apoptosis level () was increased while cell viability () was decreased in STAT3-siRNA-treated cardiomyocytes. These results together suggest that STAT3 functionally participates in the miR-320d-associated cellular activities in cardiomyocytes with AF.
Figure 5. Down-regulation of STAT3 regulated apoptosis and cell viability in cardiomyocytes with AF. (A) STAT3 mRNA expression detected by real-time PCR. (B) STAT3 protein expression detected by western blots. (C) Apoptosis levels measured by TUNEL staining assays. (D) Apoptosis level measured by Annexin V-FITC/propidium iodide (PI) assays. (E) Cell viability measured by MTT assays. Values are means ± SEM. For each experiment, sample size n ≥ 3. (*) denotes difference from control group (p < .05).
Discussion
As the most common cardiac arrhythmia, AF affects a wide range of populations worldwide [Citation1]. The occurrence of AF is becoming increasingly prevalent with higher incidence as age increases, leading to a greater number of medical and social costs, including high mortality, frequent hospitalizations, low life quality and substantial health care expenditures [Citation2,Citation31]. Therefore, there is an urgent need to explore the mechanisms underlying AF and develop new therapies to prevent and treat AF. In this study, miR-320d was decreased in cardiomyocytes with AF, causing elevated apoptosis and suppressed cell viability. Delivery of miR-320 mimics via engineered AMSCs-derived exosomes increased miR-320d expression and reversed its effect on cardiomyocytes. Also, STAT3 was a direct target of miR-320d, mediating the functions of miR-320d in cardiomyocytes with AF.
MiR-320d is a relatively less-studied member of the miR-320 family. Early studies report that the aberrant expression of miR-320d was found in various cancer cell lines and tissues, such as acute myeloid leukemia [Citation32], breast cancer [Citation33] and colorectal cancer [Citation34]. In the present study, the increased expression of miR-320d was detected in cardiomyocytes with AF and was associated with elevated apoptosis and inhibited cell viability. This is consistent with previous studies regarding the role of miR-320d in apoptosis. Qin et al. [Citation23] reported that miR-320d is a biomarker for prognosis in glioma tissues and cells and the up-regulation of miR-320d is associated with enhanced apoptosis in glioma tissues and cell lines via inhibiting the matrix metalloproteinase family. Moreover, norcantharidin induces miR-320d-dependent apoptosis in prostate cancer cells via targeting leukemia-1 (Mcl-1) [Citation35]. Collectively, miR-320d may play an important regulatory role in apoptosis in various cell types and may be a promising target to modulate apoptosis.
One of the primary goals in this study was to determine whether AMSCs-exosome was an effective delivery system to transport miR-320d to cardiomyocytes, thereby affecting cardiomyocyte activities. One important way of intercellular communication is the delivery of functional molecules via exosomes which has been widely applied to modulate cellular activities, thereby regulating disease progression and rehabilitation [Citation14]. For example, MSCs-originated exosomes enriched with miRNA-let7c attenuates renal fibrosis [Citation36]. Also, exosomes with forced expression of miR-146 leads to increased survival rate and M2 polarization in mouse sepsis [Citation37]. In the present study, miR-320d mimics were transfected into AMSCs and exosomes, which then significantly decreased miR-320d expression in cardiomyocytes with AF. Notably, cardiomyocytes co-cultured with miR-320d mimics-modified exosomes expressed opposite cell activities, including reduced apoptosis and increased viability. These results suggest that AMSCs-derived exosomes were an efficient way to deliver functional molecules to impact the cardiomyocyte activity, providing a potential therapeutic approach for AF.
Apoptosis-associated cardiomyocyte death has been extensively studied in various cardiovascular diseases, including postnatal cardiogenesis, idiopathic dilated cardiomyopathy, arrhythmogenic ventricular dysplasia and heart failure [Citation38]; however, the effects of apoptosis on AF needs to be further elucidated. In this study, the downstream signalling underlying the roles of miR-320d in cardiomyocyte apoptosis with AF were further explored. The results revealed that STAT3 was the direct target of miR-320d and the inhibition of STAT3 could reverse the effect of miR-320d in cardiomyocytes with AF. Moreover, STAT3 is activated in a context-dependent pattern in various biological processes, such as proliferation, development, differentiation and apoptosis [Citation39]. With regard to AF, STAT3 is up-regulated which mediates the structural and electrical remodelling in the rapid pacing-induced AF model [Citation40]. Activation of STAT3 signalling is also reported to contribute to the inflammation process of AF [Citation41]. Taken all together, the enhanced STAT3 expression may result from the down-regulation of miR-320d in AF, suggesting a potential signalling pathway to suppress the effect of STAT3 in AF.
In conclusion, the results in the present study demonstrated that down-regulated miR-320d promoted cardiomyocyte apoptosis and inhibited cell viability through targeting STAT3. Delivery of miR-320d mimics via AMSC-exosome reversed the effects of miR-320d on cardiomyocytes with AF. Therefore, this study may provide new insights into the mechanism underlying AF and provide a promising approach to modulate cardiomyocyte activity via exosome-mediated intercellular communication.
Author contributions
Lina Liu and Haoran Zhang conducted experiments; Lina Liu and Hongyu Mao analyzed data; Lina Liu wrote the manuscript; Xiaohong Li reviewed manuscript; Lina Liu edited the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Heeringa J, van der Kuip DA, Hofman A, et al. Prevalence, incidence and lifetime risk of atrial fibrillation: the Rotterdam study. Eur Heart J. 2006;27(8):949–953.
- De Jong AM, Maass AH, Oberdorf-Maass SU, et al. Mechanisms of atrial structural changes caused by stretch occurring before and during early atrial fibrillation. Cardiovasc Res. 2011;89(4):754–765.
- Camm AJ, Kirchhof P, Lip GY, et al. Guidelines for the management of atrial fibrillation: the Task Force for the Management of Atrial Fibrillation of the European Society of Cardiology (ESC). Eur Heart J. 2010;31(19):2369–2429.
- Nattel S. New ideas about atrial fibrillation 50 years on. Nature. 2002;415(6868):219–226.
- Benjamin EJ, Levy D, Vaziri SM, et al. Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Heart Study. JAMA. 1994;271(11):840–844.
- Ausma J, Wijffels M, Thone F, et al. Structural changes of atrial myocardium due to sustained atrial fibrillation in the goat. Circulation. 1997;96(9):3157–3163.
- Wijffels MC, Kirchhof CJ, Dorland R, et al. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation. 1995;92(7):1954–1968.
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407(6805):770–776.
- Ke L, Qi XY, Dijkhuis AJ, et al. Calpain mediates cardiac troponin degradation and contractile dysfunction in atrial fibrillation. J Mol Cell Cardiol. 2008;45(5):685–693.
- Li D, Fareh S, Leung TK, et al. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation. 1999;100(1):87–95.
- Schoonderwoerd BA, Ausma J, Crijns HJ, et al. Atrial ultrastructural changes during experimental atrial tachycardia depend on high ventricular rate. J Cardiovasc Electrophysiol. 2004;15(10):1167–1174.
- Zomer A, Vendrig T, Hopmans ES, et al. Exosomes: fit to deliver small RNA. Commun Integr Biol. 2010;3(5):447–450.
- Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, et al. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci USA. 2010;107(14):6328–6333.
- Zhang X, Yuan X, Shi H, et al. Exosomes in cancer: small particle, big player. J Hematol Oncol. 2015;8:83.
- O’Loughlin AJ, Woffindale CA, Wood MJ. Exosomes and the emerging field of exosome-based gene therapy. Curr Gene Ther. 2012;12(4):262–274.
- Yeo RW, Lai RC, Zhang B, et al. Mesenchymal stem cell: an efficient mass producer of exosomes for drug delivery. Adv Drug Deliv Rev. 2013;65(3):336–341.
- Qu Y, Zhang Q, Cai X, et al. Exosomes derived from miR-181-5p-modified adipose-derived mesenchymal stem cells prevent liver fibrosis via autophagy activation. J Cell Mol Med. 2017;21(10):2491–2502.
- Lou G, Song X, Yang F, et al. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J Hematol Oncol. 2015;8:122.
- Flynt AS, Lai EC. Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat Rev Genet. 2008;9(11):831–842.
- Cai Y, Yu X, Hu S, et al. A brief review on the mechanisms of miRNA regulation. Genomics Proteomics Bioinformatics. 2009;7(4):147–154.
- van den Berg NWE, Kawasaki M, Berger WR, et al. MicroRNAs in atrial fibrillation: from expression signatures to functional implications. Cardiovasc Drugs Ther. 2017;31(3):345–365.
- Matkovich SJ, Wang W, Tu Y, et al. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res. 2010;106(1):166–175.
- Qin CZ, Lv QL, Yang YT, et al. Downregulation of MicroRNA-320d predicts poor overall survival and promotes the growth and invasive abilities in glioma. Chem Biol Drug Des. 2017;89(5):806–814.
- Shen H, Lu S, Dong L, et al. hsa-miR-320d and hsa-miR-582, miRNA biomarkers of aortic dissection, regulate apoptosis of vascular smooth muscle cells. J Cardiovasc Pharmacol. 2018;71(5):275–282.
- Lu Y, Zhang Y, Wang N, et al. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation. 2010;122(23):2378–2387.
- Liu Y, Yan X, Sun Z, et al. Flk-1+ adipose-derived mesenchymal stem cells differentiate into skeletal muscle satellite cells and ameliorate muscular dystrophy in mdx mice. Stem Cells Dev. 2007;16(5):695–706.
- Alicka M, Marycz K. The effect of chronic inflammation and oxidative and endoplasmic reticulum stress in the course of metabolic syndrome and its therapy. Stem Cells Int. 2018;2018:4274361.
- Ullah M, Stich S, Notter M, et al. Transdifferentiation of mesenchymal stem cells-derived adipogenic-differentiated cells into osteogenic- or chondrogenic-differentiated cells proceeds via dedifferentiation and have a correlation with cell cycle arresting and driving genes. Differentiation. 2013;85(3):78–90.
- Stickney Z, Losacco J, McDevitt S, et al. Development of exosome surface display technology in living human cells. Biochem Biophys Res Commun. 2016;472(1):53–59.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25(4):402–408.
- Ho KK, Pinsky JL, Kannel WB, et al. The epidemiology of heart failure: the Framingham Study. J Am Coll Cardiol. 1993;22(4):6A–13A.
- Zhi F, Cao X, Xie X, et al. Identification of circulating microRNAs as potential biomarkers for detecting acute myeloid leukemia. PLoS One. 2013;8(2):e56718.
- Cava C, Bertoli G, Ripamonti M, et al. Integration of mRNA expression profile, copy number alterations, and microRNA expression levels in breast cancer to improve grade definition. PLoS One. 2014;9(5):e97681.
- Neerincx M, Sie DL, van de Wiel MA, et al. MiR expression profiles of paired primary colorectal cancer and metastases by next-generation sequencing. Oncogenesis. 2015;4(10):e170.
- Lin CL, Chen CM, Lin CL, et al. Norcantharidin induces mitochondrial-dependent apoptosis through Mcl-1 inhibition in human prostate cancer cells. Biochim Biophys Acta Mol Cell Res. 2017;1864(10):1867–1876.
- Wang B, Yao K, Huuskes BM, et al. Mesenchymal stem cells deliver exogenous microRNA-let7c via exosomes to attenuate renal fibrosis. Mol Ther. 2016;24(7):1290–1301.
- Song Y, Dou H, Li X, et al. Exosomal miR-146a contributes to the enhanced therapeutic efficacy of interleukin-1β-primed mesenchymal stem cells against sepsis. Stem Cells. 2017;35(5):1208–1221.
- Dispersyn GD, Ausma J, Thone F, et al. Cardiomyocyte remodelling during myocardial hibernation and atrial fibrillation: prelude to apoptosis. Cardiovasc Res. 1999;43(4):947–957.
- Al Zaid Siddiquee K, Turkson J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008;18(2):254–267.
- Tsai CT, Lin JL, Lai LP, et al. Membrane translocation of small GTPase Rac1 and activation of STAT1 and STAT3 in pacing-induced sustained atrial fibrillation. Heart Rhythm. 2008;5(9):1285–1293.
- Jiang Q, Ni B, Shi J, et al. Down-regulation of ATBF1 activates STAT3 signaling via PIAS3 in pacing-induced HL-1 atrial myocytes. Biochem Biophys Res Commun. 2014;449(3):278–283.