
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Ion-complementary self-assembling peptides have potential in delivering hydrophobic drugs. This study involved two self-assembling peptides, RADA16-I and RVDV16-I, of which RVDV16-I was a novel self-assembling peptide with different hydrophobic side chains designed from RADA16-I. The purpose of this study was to observe the interaction between different self-assembling peptides and emodin through fluorescence spectrophotometry, CD, SEM and AFM; to construct a preliminary suspension in-situ hydrogel delivery system for emodin with the self-assembling peptides; and to investigate the drug-loading and drug-releasing properties of the self-assembling peptides on emodin. The results showed that both peptides can interact with emodin and the interaction was dominated by hydrophobic interaction. The aqueous solutions of both self-assembling peptides can form relatively stable suspensions with emodin under mechanical stirring, and the suspension can form in-situ hydrogel under physiological condition. In vitro release of emodin from the hydrogels showed a manner of sustained release to some extent. Cell viability studies showed inherent proliferation inhibiting effects of emodin on tumor cells was maintained or enhanced through the in-situ hydrogels. The self-assembling peptides RADA16-I and RVDV16-I had showed promising drug-loading and drug-releasing performance for hydrophobic drugs. It is reasonable to exploit self-assembling peptides as drug carriers for their great potential to improve delivery of hydrophobic drugs.
Introduction
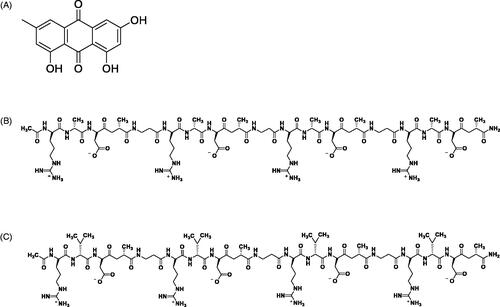
Emodin (1,3,8-trihydroxy-6-methylanthraquinone, EM), with formula of C15H10O5, shown in , is one of the major bioactive anthraquinones existed in the Radix et Rhizoma Rhei and other medical plants. It has a wide range of pharmacological activities including bacteriostatic effect, anti-inflammation, protection of liver and kidney, improvement of blood microcirculation, and anti-tumor effect [Citation1–4]. Many scholars have conducted in-depth researches and found that emodin has inhibitory effects on solid tumors such as ovarian cancer [Citation5], breast cancer [Citation6], lung cancer [Citation7–10], liver cancer [Citation11–13], and non-solid tumors such as leukemia [Citation14–16]. However, poor water solubility and possible sudden release after administration made emodin not conducive to injection or oral administration. Even in in vitro and in vivo experiments, the concentration of emodin is limited due to the need of organic solvents such as dimethyl sulfoxide in the dissolution operation. The formulations such as liposomes, solid dispersions, nanoemulsions, and nanomicelles can partly resolve the water solubility problem [Citation17–19]. Unfortunately, due to low drug loading and complicated preparation process of these formulations, also the poor biocompatibility and biodegradability of the polymer material used as drug carriers in these formulations, it is still a major challenge in the field of pharmaceutical preparations to find an ideal drug carrier with good biocompatibility and high drug loading to achieve effective delivery release of hydrophobic drugs.
Figure 1. Chemical structures of (A) emodin (B) RADA16-I (C) RVDV16-I.
With environment-sensitive properties similar to natural extracellular matrices, hydrogels are widely used in biomedical field. Especially, the in situ hydrogel system usually made from polymer materials can undergo phase change immediately after administration and reversibly transform from a solution into a semi-solid or solid gel state [Citation20,Citation21]. This property imparts injectability to in situ hydrogel system and makes it suitable for local and minimally invasive drug delivery, while hydrophobic drugs can be accommodated into the hydrophobic regions of the three-dimensional network of nanofiber structures of the hydrogels. Thus, hydrophobic drugs can be administered locally through drug-loaded in situ hydrogel system and then slowly released from in situ hydrogels into wanted tissues by degradation of the hydrogels [Citation22].
In recent years, self-assembling peptides have been widely researched as nanomaterials in cell culture [Citation23,Citation24], tissue engineering, organ regeneration repairing [Citation25–28], hemostasis [Citation29,Citation30], drug delivery [Citation31–35] and other biomedical fields. Self-assembling peptides include ion-complementary self-assembling peptides, amphiphilic peptides, cyclic peptides, surface-active peptides, aromatic peptides, and many other types [Citation36]. Among them, ion-complementary self-assembling peptides have attracted the most research interests of many researchers. The ion-complementary self-assembling peptides contain alternately arranging positively charged amino acids and negatively charged amino acids with hydrophobic amino acids in their structures, and thus can self-assemble under the electrostatic force, hydrophobic interaction and hydrogen bond [Citation37]. Studies have shown that the ion-complementary self-assembling peptide RADA16-I (shown in ) is not only a potential carrier for protein drugs [Citation20], but also can pack strong anticancer activity of the drug paclitaxel [Citation38] and 5-fluorouracil [Citation39]. The potential of ion-complementary self-assembling peptides as carrier of hydrophobic drugs need to be exploited and verified through further subsequent researches.
The purpose of this study was to develop a suspension-in situ hydrogel drug delivery system of emodin based on self-assembling peptide RADA16-I and RVDV16-I. The latter is a novel self-assembling peptide (shown in ), and it was designed and synthesized by replacing hydrophobic amino acid alanin (A) in RADA16-I with more hydrophobic valine (V). In this study, the interactions between the self-assembling peptides and emodin, formation and part of the characteristics of the suspension-in situ hydrogel drug delivery system, the controlled release characteristics and anti-tumor effects of emodin from the in situ hydrogels were studied by fluorescence analysis, particle size distribution, morphological analysis, rheological tests, in vitro release studies, and cell viability experiments.
Materials
Emodin reference (Batch No. 110758–200110) and emodin raw materials (Batch No. ZL20140928), with purity of emodin both greater than or equal to 98%, were purchased from Guizhou Dida Biotechnology Co., Ltd (Guizhou, China) and Nanjing Zelang Pharmaceutical Technology Co., Ltd (Nanjing, China), respectively. High glucose Dulbecco’s Modified Eagle’s Medium and 1% antibiotics (100 × streptomycin-penicillin) were purchased from Hyclone Inc. (Logan, UT, USA). 3–(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) and phosphate buffer saline (PBS) were purchased from Beijing Solarbio Technology Co., Ltd (Beijing, China). Dimethyl sulfoxide (DMSO) was purchased from Beijing Dingguo Changsheng Biotechnology Co., Ltd (Beijing, China). Fetal bovine serum (FBS) was purchased from Gibco (Thermo Fisher Scientific, Inc., Waltham, MA, USA). 0.25% Trypsin-EDTA solution was purchased from Biosharp Inc. (Shanghai, China). Human cancer cell lines: A549 (human lung carcinoma cell line) and HepG2 (Human hepatoma cell line) were kindly donated by the Department of Biochemistry of Zunyi Medical University. Cell culture bottles and other related consumables were purchased from Corning, Inc. (Corning, NY, USA).
The self-assembling peptides RADA16-I (1712.77 g/mol) and RVDV16-I (1937.20 g/mol) were commercially synthesized by Shanghai Bootech Bioscience & technology Co., Ltd (Shanghai, China). The peptides comprise the amino acid sequences n-RADARADARADARADA-c for RADA16-I and n-RVDVRVDVRVDVRVDV-c for RVDV16-I, where R is for arginine, A is alanine, D is aspartic acid, and V is valine. The N terminus and C terminus of the peptides were protected by acetyl and amino groups, respectively.
Methods
Physical and chemical properties of emodin
In this experiment, the solubility of emodin in some related solvents was determined by the equilibrium method [Citation40,Citation41]. The emodin reference substance and the test solution were prepared by using anhydrous ethanol. The solutions and the peptide solutions being scanned in a wavelength range of 200–400 nm of ultraviolet spectrophotometer (T9CS, Beijing, China). A series of concentration of reference solutions were used to measure the absorbance, and linear regression was performed with absorbance and concentration. And the solubility of emodin in pure water, pH 7.4 and pH 6.8 PBS, ethanol and methanol were calculated by standard curve.
Formation of peptide-emodin suspensions
Both ion-complimentary self-assembling peptide solutions (3–10 mg/mL) were prepared by dissolving peptides powder in Milli-Q water with 2 min vortex, followed by sonication for 10 min to dissolve the peptides in water completely. The peptide solutions were maintained at 4 °C before performing characterization. To prepare peptide-emodin (EM) suspensions, proper amount of 3, 5, 7 or 10 mg/mL peptide solutions were added into glass vials with EM all at a final concentration of 1 mg/mL, followed by magnetic stirring at 1000 r/min and room temperature for 48 h before characterization and cytotoxicity studies. All of the vials and solvents were sterilized, and the samples were prepared in a biological safety cabinet to avoid possible contamination for cell culture experiments.
Fluorescence spectroscopy measurement of emodin
Proper amount of EM in ethanol (1 mg/mL, 24 μL) was added to several vials and the ethanol was volatilized in the dark to obtain EM films. Then, 4 mL of 10–200 μM series of RADA16-I and RVDV16-I solutions were added to the films, respectively. The mixture was sonicated for 3 h at room temperature and allowed to stand for 24 h in the dark. With an aqueous solution containing no peptide as control, the emission spectra of EM in the solutions were recorded.
4 mL of 100 μM RADA16-I and RVDV16-I aqueous solutions were added to EM films similarly prepared as mentioned above and the emission spectra of EM in the solutions were recorded immediately after the solutions being magnetically stirred for 0.5, 2, 4, 8, 12, 24, 36, 48, 60, 72 h with an aqueous solution containing no peptide as control.
The fluorescence intensity of EM in the peptide solutions were detected by fluorescence spectrophotometer (F-380, Tianjin, China) with an excitation and emission spectral slits width of 5 nm. The excitation wavelength was set as 254 nm, the emission wavelength range 400–600 nm, the PMT voltage 700 V, the scanning speed 1200 nm/min, and the response time 0.05 s. The maximum fluorescence emission intensity of EM at the emission wavelength of 509 nm was recorded, and the changes of fluorescence intensity of EM in the peptide solutions with time were observed.
Circular dichroism (CD)
In this experiment, a circular dichroism spectrometer (V100, Applied Photophysics, UK) was used to detect the secondary structure of the two peptides without and with EM. Measurement conditions were 190–260 nm wavelength, 1 mm cuvette optical path, 1 mm resolution, and 100 nm/min scan speed. The baseline correction was subtracted from all spectra and the data was expressed as the molar ellipticity in deg.cm2.dmol/L.
Atomic force microscopy (AFM)
Peptide solutions without and with EM at designed peptide concentration were chosen for AFM imaging. The appropriate amounts of EM powder were dissolved in ethanol and appropriate amount of the solutions were transferred into a vial, then a layer of EM film was obtained by evaporating the ethanol. Peptide solutions at a concentration of 200 μM were added to the films and the final concentration of EM was 0.1 μM. 10 μL of peptide solutions without and with EM were dropped onto the newly exfoliated mica plate, allowed to stand for 60 s. The samples were adhered to the surface of the mica plate with the unabsorbed parts removed using the Milli-Q water, allowed to dry naturally at room temperature or blown to dry with nitrogen. The nanostructure of the sample was detected in a tapping mode on atomic force microscope (Dimension ICON, Bruker, USA).
Dynamic light scattering
Peptide-EM suspensions were made at the peptide-to-EM ratio of 1:1, 2:1, 5:1, respectively. The average particle size and particle size distribution in the suspensions were detected on a particle size analyzer (90 Plus, Brookhaven Ltd, USA).
Scanning electron microscope (SEM)
Surface morphology of the particles in peptide-EM suspension and peptide-EM in situ hydrogel scaffold were analyzed by scanning electron microscope (SEM). For suspension, 10 μL of each suspension on a silicon wafer and air dry at room temperature and then coated with gold before the measurement, imaged, and photographed by SEM (TESCAN MIRA 3, Taizhou, China) at an acceleration voltage of 10.00 kV; For in situ hydrogel, the peptide solutions or the suspensions were dropped into PBS to form hydrogel, the hydrogel was first soaked with glutaraldehyde and immersed it in Milli-Q water, then used a series of concentrations (50%–100%) of ethanol solution for gradient dehydration, and the sample was dried by CO2 critical point dryer. After the sample was completely dried, the sample was adhered to a metal table for gold spraying, and then the sample was scanned by SEM (S-4800, Hitachi, Japan).
Rheological analysis
The rheological properties of peptide solutions and peptide-EM suspensions were measured on a Rheometer (DHR-3, TA Instruments, New York, USA). Self-assembling peptide-EM suspension was prepared with the final concentration of EM 0.5 mg/mL and the final concentrations of RADA16-I and RVDV16-I 3.0, 5.0 and 7.0 mg/mL, respectively. Each 80 μL sample of the suspensions were placed on the plate of the rheometer and a trap was placed around the cone to seal the liquid. The storage modulus (G′) and loss modulus (G″) was measured at 25 °C for the frequency sweep tests with strain of 0.5% and frequency range from 0.1 to 10.0 rad/s. For rheological testing of peptide and peptide-EM in situ hydrogel, peptide solutions and peptide-EM suspensions were first induced to hydrogel by adding 40 μL of phosphate-buffered saline solution. After equilibrating for 10 min, the excessive solution was taken off, and the measurement was performed as described above.
In vitro release measurement
The release of EM from peptide-EM hydrogel was carried out in vitro in phosphate-buffered saline solution (PBS). 600 μL of pH 7.4 PBS solution and 600 μL of peptide-EM suspension were mixed in a dialysis bag. After equilibrating for 10 min, the dialysis bag was tied. Then, the dialysis bag was placed in a centrifuge tube containing 30 mL of release medium. The same volume of EM and water were added into the dialysis bag as the control of free EM. In vitro release test was carried out by shaking the centrifuge tubes at 100 r/min in 37°C. 3 mL of release medium was taken out at the set time points (0, 2, 4, 6, 8, 12, 24, 30, 36, 48, 72 h) and the same volume of fresh release medium was immediately replenished to ensure that all tests were done under leaky conditions. The UV absorbance of the release samples filtered through a 0.45 μm filter was measured at wavelength of 254 nm to calculate the cumulative release rates of EM from the self-assembling peptide hydrogels. Cumulative release rate was calculated by the following equation:
Q represents the cumulative release rate of EM, Cn represents the concentration of EM at the time of sampling, Vn represents the volume of each sample, Cn−1 represents the concentration of EM at the previous sampling point, V represents the volume of the release medium, and D represents the mass of EM in the hydrogels (or the suspensions).
In vitro cell viability studies
A549 and HepG2 cells were cultured in a 96-well plate at a density of 2 × 104 cells/mL and incubated for 24 h. Cells were treated with EM aqueous suspension and in situ peptide-EM hydrogels (formed from peptide-EM suspensions in cell culture media) at different concentrations of EM (20, 40, 60, 80, 100, 120, 140 and 160 μM) and incubated for 24 and 48 h at 37 °C. Untreated cells incubated in medium and the medium served as control and blank, respectively. After incubation, MTT (20 μL/well) was added into each well in a final concentration of 5 mg/mL. Cells were further incubated with MTT for 4 h. The insoluble formazan was dissolved in dimethyl sulfoxide (150 μL/well) solution. The plate was further shaken for 10 min on a shaker at room temperature. Cell viability was assessed by measuring absorbance at 490 nm using a 96-plate reader (Bio-Rad Laboratories, Hercules, CA, USA). In addition, the morphology of individual groups of cells after the 48 h treatment with EM was observed using microscope (Olympus, Tokyo, Japan).
Statistical analysis
The statistical analysis was performed using analysis of One-way ANOVA and Student’s t-test (SPSS 20.0 statistical software). All data were expressed as Mean ± standard deviation (SD), and probability (p) smaller than .05 was considered that the difference was statistically significant.
Results and discussion
Physical and chemical properties of emodin
As the related literatures have reported, the apparent oil-water partition coefficient of EM is about 1.06 [Citation42], and the dissociation constant pKa is about 8.25 [Citation43], indicating that EM is a weakly acidic drug. From the results of wavelength scanning in , in the range of 200–400 nm, EM had three ultraviolet absorption peaks at 220 nm, 254 nm and 298 nm, with the maximum at 220 nm, while ethanol had no obvious absorption. So 220 nm was chosen as the detection wavelength for determination of EM. The standard curve equation for EM was C = 0.0940A + 0.0101 (r2 = 0.9993). It can be seen from that solubilities of EM in organic solvents were much higher than those in aqueous solutions, and EM could be defined as an almost insoluble drug [Citation44].
Figure 2. The UV spectrum of EM.
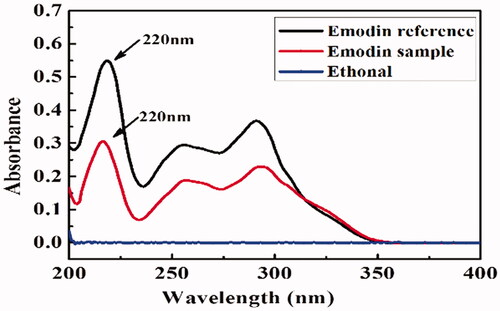
Table 1. Solubility of EM at different medium.
Peptide design
As a classical ion-complementary self-assembling peptide, RADA16-I can assemble to form a β-folded secondary structure and form nanofibers, thus it has been widely used in various research fields. There are a hydrophilic surface with positively charged arginine (Arg, R) and negatively charged aspartic acid (Asp, D) and a hydrophobic surface with non-polar amino acid alanine (Ala, A) in the structure of RADA16-I [Citation45,Citation46]. RADA16-I has shown some potentials to be carriers of hydrophobic compounds or drugs for its hydrophobic side can interact with a hydrophobic compounds or drugs, while its hydrophilic side can make the hydrophobic compound or drug relatively stable in aqueous solution systems [Citation47,Citation48]. In the purpose of exploring whether alteration of non-polar amino acid can change the encapsulation and release performance of ion-complementary self-assembling peptides on hydrophobic drugs, RVDV16-I was designed by replacing the hydrophobic amino acid alanine (A) with more hydrophobic valine (V) based on the structure of RADA16-I. The sequences and characteristics of the two peptides were listed in .
Table 2. The sequences and characteristics of the two peptides.
Interaction between peptides and emodin
Fluorescence of EM with the self-assembling peptides
The fluorescent probe method is commonly used in determining the aggregation state, stability, and micellization behavior of polymer in aqueous solutions. This method was adopted to investigate the interactions between EM and self-assembling peptide RADA16-I or RVDV16-I by making use of steady-state fluorescent response of EM to the microenvironment [Citation49]. The steady-state fluorescence emission spectra of EM in aqueous solutions of self-assembling peptides and in water under mechanically stirring were shown in . During EM being mechanically stirred with pure water for 72 h, the fluorescence spectra of EM were disordered and irregular, and the fluorescence intensities were extremely weak (). While, the fluorescence intensities of EM in the two peptide solutions increased with the increase of stirring time. After EM in self-assembling peptide aqueous solutions were stirred for a certain time, which can be regarded as the stabilization time of EM stirred in the peptides’ aqueous solutions, 24 h for RADA16-I and 36 h for RVDV16-I, the fluorescence intensity tended to be unchanged stable. The longer stabilization time for EM in RVDV16-I aqueous solution may be related to the higher hydrophobicity of RVDV16-I than that of RADA16-I, and encapsulating a larger amount of EM required more time to stabilize them in aqueous solutions. As shown in , after the same concentration of EM interacted with two peptides at a series of concentrations, the fluorescence pattern was similar. The fluorescence intensity of EM increased with the increased concentration of peptides. It was noted that the fluorescence intensity of EM in RVDV16-I solutions were significantly higher than those in RADA16-I solutions at the same peptide concentration of 50 or 100 μM. This may indicate stronger hydrophobic interaction of RVDV16-I with hydrophobic compounds than that of RADA16-I.
Figure 3. Emission spectra of EM at the excitation wavelength of 254 nm in self-assembling peptide aqueous solutions and in water under mechanically stirring. (A) RADA16-I; (B) RVDV16-I; (C) Water; (D) Fluorescence intensity of EM at 509 nm; [EM] = 6.0 μg/mL, [RADA16-I] = [RVDV16-I] = 100 μM.
![Figure 3. Emission spectra of EM at the excitation wavelength of 254 nm in self-assembling peptide aqueous solutions and in water under mechanically stirring. (A) RADA16-I; (B) RVDV16-I; (C) Water; (D) Fluorescence intensity of EM at 509 nm; [EM] = 6.0 μg/mL, [RADA16-I] = [RVDV16-I] = 100 μM.](/cms/asset/a77e4ec4-2cb3-4bd8-90b4-b14e447508a5/ianb_a_1673768_f0003_c.jpg)
Figure 4. Fluorescence spectra of EM in different concentration of RADA16-I and RVDV16-I. (A) RADA16-I; (B) RVDV16-I. [EM] = 6.0 μg/mL, [RADA16-I] = [RVDV16-I] = 0–200 μM.
![Figure 4. Fluorescence spectra of EM in different concentration of RADA16-I and RVDV16-I. (A) RADA16-I; (B) RVDV16-I. [EM] = 6.0 μg/mL, [RADA16-I] = [RVDV16-I] = 0–200 μM.](/cms/asset/95e667ec-0bcb-4d07-aa49-5e6f1d73ff58/ianb_a_1673768_f0004_c.jpg)
Circular dichroism
Circular dichroism is a very important tool for studying the secondary structure of peptides or proteins. The secondary structure of the protein has four common forms: α-helix, β-sheet, β-turn and random curl. The secondary structure with β-sheet generally has a positive absorption peak at 195–197 nm and a negative absorption peak at 217–218 nm. It can be seen from that the peptides, RADA16-I and RVDV16-I, both had a typical β-sheet secondary structure; in the absence of EM, the absorption intensities increased significantly; and the absorption intensity also increased as the concentration of the peptides increased. At the same peptide concentration, the absorption intensity of RVDV16-I was lower than that of RADA16-I, which might be due to the increased surface tension caused by the stronger hydrophobic interaction of RVDV16-I with EM. These results indicated that RADA16-I and RVDV16-I both can interact with EM and the interaction can result in a slight change in the secondary structure of the peptides.
Figure 5. Secondary structures of RADA16-I, RVDV16-I, RADA16-I-EM and RVDV16-I-EM complexes. The concentrations of RADA/RVDV16-I in RADA/RVDV16-I-EM-1, RADA/RVDV16-I-EM-2, RADA/RVDV16-I-EM-3 were 0.1, 0.2, 0.4 mg/mL, respectively, [EM] = 0.05 mg/mL.
![Figure 5. Secondary structures of RADA16-I, RVDV16-I, RADA16-I-EM and RVDV16-I-EM complexes. The concentrations of RADA/RVDV16-I in RADA/RVDV16-I-EM-1, RADA/RVDV16-I-EM-2, RADA/RVDV16-I-EM-3 were 0.1, 0.2, 0.4 mg/mL, respectively, [EM] = 0.05 mg/mL.](/cms/asset/4a5f28e7-927c-4cd6-80ac-fa64eec71118/ianb_a_1673768_f0005_c.jpg)
showed the morphology RADA16-I and RVDV16-I in aqueous solutions without and with EM. It can be seen from the figure that RADA16-I and RVDV16-I can self-assemble in water to form a linear elongated nanofiber structure. In the presence of EM, though RADA16-I kept a slender fiber structure, the width and height of the fibers increased. RVDV16-I tended to form a globular and fiber-like structure, which might be related to the more phobic amino acids in the peptide sequence. Similarly, the width and height of the fibers of RVDV16-I slightly increased in the presence of EM, but the microstructure seemed to show a trend of homogenization. These phenomena indicated that RADA16-I/RVDV16-I and EM can interact with each other and EM can be encapsulated by both RADA16-I and RVDV16-I.
Figure 6. AFM images of RADA16-I, RVDV16-I, RADA16-I-EM and RVDV16-I-EM complexes in aqueous solutions. (A) RADA16-I, (B) RADA16-I-EM, (C) RVDV16-I, (D) RVDV16-I-EM, peptide concentration: [RADA16-I] = [RVDV16-I] = 200 μM, [EM] = 0.1 μM.
![Figure 6. AFM images of RADA16-I, RVDV16-I, RADA16-I-EM and RVDV16-I-EM complexes in aqueous solutions. (A) RADA16-I, (B) RADA16-I-EM, (C) RVDV16-I, (D) RVDV16-I-EM, peptide concentration: [RADA16-I] = [RVDV16-I] = 200 μM, [EM] = 0.1 μM.](/cms/asset/c9b077ab-39b0-4e15-91d4-7cced8634061/ianb_a_1673768_f0006_c.jpg)
Formation and characterization of peptide-emodin suspensions
After being magnetically stirred for 48 h, both the self-assembling peptides formed orange colloidal suspensions with EM in water. While EM was difficult to be suspended in pure water without any peptide and a large amount of EM powder attached to the stirrers and the vial wall ().
Figure 7. Formation of colloidal suspensions of EM in RADA16-Ior RVDV16-I aqueous solution. (A) In water; (B) In RADA16-I; (C) In RVDV16-I; [EM] = 1.0 mg/mL, [RADA16-I] = [RVDV16-I] = 1.0 mg/mL.
![Figure 7. Formation of colloidal suspensions of EM in RADA16-Ior RVDV16-I aqueous solution. (A) In water; (B) In RADA16-I; (C) In RVDV16-I; [EM] = 1.0 mg/mL, [RADA16-I] = [RVDV16-I] = 1.0 mg/mL.](/cms/asset/e353b23b-cbc3-4b85-90d8-cb0e99012122/ianb_a_1673768_f0007_c.jpg)
The morphology of particles in the self-assembling peptide-EM suspensions were characterized by SEM (). From , it can be seen that particles in the suspension of EM formed in pure water presented a large irregular shape; in comparison, the particles in the suspensions with 0.05 mg/mL of RADA16-I presented rod-like shaped (). It can be speculated that the hydrophobic region of EM combined with the hydrophobic region of the peptide and the large EM particles were broken under the action of magnetic stirring. When added RADA16-I peptide increased to 0.10 mg/mL, fibrous formation occurs (). When the concentration of RADA16-I peptide continues to increase to 1.0 mg/mL, a relatively regular rod shape was formed (). The shape of the particles in suspension formed by 0.05 mg/mL RVDV16-I was irregular and there was no fibrous formation in the solution (), but as the concentration of the peptide increased to 0.1 mg/mL, the morphology of the particles in the suspensions began to become regular (). At the RVDV16-I concentration of 1.0 mg/mL RVDV16-I, EM was encapsulated in peptide, and presented a particulate shape ().
Figure 8. SEM images of the peptide-EM suspensions at different concentration of RADA16-I and RVDV16-I. (A) EM aqueous suspension without peptide; (B, C, D) RADA16-I-EM; (E, F, G) RVDV16-I-EM. [EM] = 0.10 mg/mL; from left to right, peptide concentrations were 0.05, 0.1, 1.0 mg/mL, respectively.
![Figure 8. SEM images of the peptide-EM suspensions at different concentration of RADA16-I and RVDV16-I. (A) EM aqueous suspension without peptide; (B, C, D) RADA16-I-EM; (E, F, G) RVDV16-I-EM. [EM] = 0.10 mg/mL; from left to right, peptide concentrations were 0.05, 0.1, 1.0 mg/mL, respectively.](/cms/asset/d502caa4-0f52-4dd7-82ef-4b4363883c81/ianb_a_1673768_f0008_b.jpg)
Particle size distribution in suspensions of EM with and without self-assembling peptides were shown in . The suspension of EM formed in pure water had a particle size ranging from 1000 to 10000 nm with an average of 2584.9 nm. While the average particle size of the suspensions of EM formed in RADA16-I and RVDV16-I water solutions were 485.4 nm and 420.8 nm, respectively. Both were about 5 times lower than that in EM-water suspension (p < .05).
Figure 9. Particle size distribution in suspensions of EM with and without self-assembling peptides. (A) size distribution; (B) average size and PDI. [EM] = 0.5 mg/mL, [RADA16-I] = [RVDV16-I] = 1.0 mg/mL. *p < .05, average particle size vs average particle size in water suspension; #p < .05, PDI vs PDI in water suspension.
![Figure 9. Particle size distribution in suspensions of EM with and without self-assembling peptides. (A) size distribution; (B) average size and PDI. [EM] = 0.5 mg/mL, [RADA16-I] = [RVDV16-I] = 1.0 mg/mL. *p < .05, average particle size vs average particle size in water suspension; #p < .05, PDI vs PDI in water suspension.](/cms/asset/b59e20e0-3ee9-43a4-97ab-20f83cf52fef/ianb_a_1673768_f0009_c.jpg)
The suspension amount and solubility of EM in self-assembling peptide solution were shown in . The suspension rate of EM in water was 11.4%; the suspension rate in 10–50 μM peptide solution increased with the increase of peptide concentration, and the maximum suspension rate in 50 μM peptide solution was 28.0%. When the concentration of peptide increased to 100 μM, the EM suspension rate in RADA16-I and RVDV16-I solution dramatically increased to 90.57% and 92.25%, respectively. The suspension rate of EM in 100 μM peptide solution was much higher than that in water (p < .05). There was no significant difference in the suspension rate of EM between in the two peptide solutions (p > .05). The solubility of EM in peptide solutions increased with the increase of peptide concentration. The solubility of EM in 1000 μM RADA16-I and RVDV16-I solutions were 152.0 μg/mL and 173.0 μg/mL, respectively. Both were much higher than that in water (p < .05). EM was more soluble in RVDV16-I solution than RADA16-1 solution.
Figure 10. (A) The suspension rate of EM in RADA16-I and RVDV16-I solution ( ± s, n = 3). [EM] = 0.5 mg/mL, [RADA16-I] = [RVDV16-I] = 10–100 μM, (B) Solubility of EM at different concentration of RADA16-I and RVDV16-I (
± s, n = 3). [RADA16-I] = [RVDV16-I] = 100–1000 μM. *p < .05, **p < .01 vs in pure water without peptide.
![Figure 10. (A) The suspension rate of EM in RADA16-I and RVDV16-I solution (x¯ ± s, n = 3). [EM] = 0.5 mg/mL, [RADA16-I] = [RVDV16-I] = 10–100 μM, (B) Solubility of EM at different concentration of RADA16-I and RVDV16-I (x¯ ± s, n = 3). [RADA16-I] = [RVDV16-I] = 100–1000 μM. *p < .05, **p < .01 vs in pure water without peptide.](/cms/asset/5865abd6-fec4-479f-82ae-e168cc378729/ianb_a_1673768_f0010_b.jpg)
Formation and characterization of in situ hydrogels
After being magnetically stirred in water for 48 h, the self-assembling peptides and EM formed orange colloidal suspensions. Hydrogels were formed rapidly when the peptide-EM suspensions were added into either pH 7.4 PBS or physiological saline at 37 °C (). The surface morphology of the in situ hydrogels was characterized by SEM. As can be seen from , the in situ hydrogels formed from RADA16-I solution being dropped into PBS presented a fibrous network structure (). With the increase of the concentration of RADA16-I, the network structure became denser (). Compared with RADA16-I, the in situ hydrogels formed from RVDV16-I presented more compact network structure ()). This may be associated with stronger hydrophobicity of RVDV16-I. It can be seen that the morphology of the hydrogels formed from drug-loaded peptide-EM suspensions were different from that formed from corresponding peptide solution, and there were granular particles adhered to the peptide nano mesh structure. It can be reasonably speculated that EM nano- or micro- particles were dispersed and adsorbed onto nanonetworks of the assembling peptides. The SEM results showed that the expected suspension-in situ hydrogel delivery system for the hydrophobic drug EM was preliminarily established with the self-assembling property of the peptide RADA16-I or RVDV16-I. This also further demonstrated that the self-assembling peptide can interact with hydrophobic drug and can be developed as a carrier for hydrophobic drugs.
Figure 11. Formation of in situ hydrogel of RADA16-I and RVDV16-I-EM suspension at PBS and 0.9% NaCl solution; (A) RADA16-EM suspension in PBS; (B) RADA16-I-EM suspension in 0.9% NaCl; (C) RVDV16-I-EM suspension in PBS; (D) RVDV16-I-EM suspension in 0.9% NaCl; [EM] = 1.0 mg/mL, [RADA16-I] = [RVDV16-I] = 5.0 mg/mL.
![Figure 11. Formation of in situ hydrogel of RADA16-I and RVDV16-I-EM suspension at PBS and 0.9% NaCl solution; (A) RADA16-EM suspension in PBS; (B) RADA16-I-EM suspension in 0.9% NaCl; (C) RVDV16-I-EM suspension in PBS; (D) RVDV16-I-EM suspension in 0.9% NaCl; [EM] = 1.0 mg/mL, [RADA16-I] = [RVDV16-I] = 5.0 mg/mL.](/cms/asset/ee219ab1-771e-4f58-b618-100fd844d6f3/ianb_a_1673768_f0011_c.jpg)
Figure 12. SEM images of in situ hydrogels when the peptide solution or the peptide-EM suspension was dropped into pH 7.4 PBS. (A–C) RADA16-I, (D–F) RADA16-I-EM, (G–I) RVDV16-I, (J–L) RVDV16-I-EM, peptide concentration: from left to right were 5, 10, 20 mg/mL, [EM] = 0.5 mg/mL.
![Figure 12. SEM images of in situ hydrogels when the peptide solution or the peptide-EM suspension was dropped into pH 7.4 PBS. (A–C) RADA16-I, (D–F) RADA16-I-EM, (G–I) RVDV16-I, (J–L) RVDV16-I-EM, peptide concentration: from left to right were 5, 10, 20 mg/mL, [EM] = 0.5 mg/mL.](/cms/asset/954c0069-2a17-43ae-8871-b3d9e784ea5d/ianb_a_1673768_f0012_b.jpg)
The gelation of peptide-EM suspension was characterized by rheometry at 37 °C, and taking pH 7.4 PBS to simulate body fluid. As shown in , in the RADA16-I water solution, the G' of the solution was slightly larger than G'', but all the values were under 10 Pa, indicating no hydrogel formed in the pure RADA16-I water solution. When the peptide solution was dropped into PBS, G' and G'' increased by nearly 10 times, and G′ > G″, indicated that hydrogels formed in situ (). The G' of the RVDV16-I water solution was over 10 Pa, and G′ and G″ increased the same as that of RADA16-1 after mixing with PBS, indicating hydrogel formed (). The results in C and D of indicated that the suspension formation by self-assembling peptides interacting with EM basically did not affect the gelation behavior of the peptides with G′ and G″ remained consistent or slightly enhanced. As shown in , as the concentration of the peptide increased, the G′ and G″ of the hydrogel increased, and G′ was always larger than G″. The frequency sweep results showed that both G′ and G″ of the hydrogel remained stable when the shear frequency changed from 0.1 to 10.0 rad./s, while the G′ and G″ values of the peptide solution or the peptide-EM suspension fluctuated with increased shear frequency. The results from rheometry study suggested that the suspension-in situ hydrogel delivery system for the hydrophobic drug EM was successfully established, though the hydrogel strength may still need to be improved.
Figure 13. Rheological properties of RADA16-I, RVDV16-I, RADA16-I-EM and RVDV16-I-EM solution (or suspension) and hydrogels. (A) RADA16-I; (B) RVDV16-I; (C) RADA16-I-EM; (D) RVDV16-I-EM. [EM] = 1.0 mg/mL, [RADA16-I] = [RVDV16-I] = 5 mg/mL.
![Figure 13. Rheological properties of RADA16-I, RVDV16-I, RADA16-I-EM and RVDV16-I-EM solution (or suspension) and hydrogels. (A) RADA16-I; (B) RVDV16-I; (C) RADA16-I-EM; (D) RVDV16-I-EM. [EM] = 1.0 mg/mL, [RADA16-I] = [RVDV16-I] = 5 mg/mL.](/cms/asset/adaa1637-4a29-4c3c-8e79-e644dd528d9b/ianb_a_1673768_f0013_b.jpg)
Figure 14. Rheological properties of peptide-EM hydrogels formed from peptide-EM suspensions with different concentration of RADA16-I and RVDV16-I (A) RADA16-I-EM hydrogel; (B) RVDV16-I-EM hydrogel; (▲, △): 3 mg/mL peptide; (●, ^): 5 mg/mL peptide; (■, □): 7 mg/mL peptide; [EM] = 1.0 mg/mL, [RADA16-I] = [RVDV16-I] = 3, 5, 7 mg/mL.
![Figure 14. Rheological properties of peptide-EM hydrogels formed from peptide-EM suspensions with different concentration of RADA16-I and RVDV16-I (A) RADA16-I-EM hydrogel; (B) RVDV16-I-EM hydrogel; (▲, △): 3 mg/mL peptide; (●, ^): 5 mg/mL peptide; (■, □): 7 mg/mL peptide; [EM] = 1.0 mg/mL, [RADA16-I] = [RVDV16-I] = 3, 5, 7 mg/mL.](/cms/asset/8a9bd2d4-d598-4ede-9f24-e707058e28b7/ianb_a_1673768_f0014_b.jpg)
In vitro EM release from peptide-EM hydrogel
As shown in , EM was released fast from aqueous suspension without peptides with a significant burst release, about 20% of EM was released in 2 h, and the cumulative release at 36 h was close to 90%. In the in situ hydrogel formed from 3 mg/mL of RADA16-I-EM suspension, about 20% of EM was released at 2 h with a slight burst release, and the cumulative release of EM was about 80% at 48 h. No obvious burst release was observed in RADA16-I hydrogels at concentrations of 5 mg/mL and 7 mg/mL and the cumulative release of EM from the in situ hydrogels formed from 5, 7 mg/mL of RADA16-I -EM suspension, at 84 h was 87.36%, and 83.81%, respectively. There was no burst release in the in situ hydrogel formed from RVDV16-I-EM suspensions and the cumulative release of EM from the in situ hydrogels formed from 3, 5, 7 mg/mL of RVDV16-I-EM suspension was 89.12%, 86.42%, and 80.46%, respectively. In vitro release results showed that the in situ hydrogel formed from self-assembling peptide -EM had a sustained release effect to some extent, and the release of EM became slower as the concentration of peptide increased. It was noted that the hydrogel formed from RVDV16-I -EM suspension had a cumulative release rate slightly lower than that of RADA16-I. This release experiment initially confirmed the feasibility of constructing a suspension-in situ hydrogel carrier system for hydrophobic drugs by self-assembling peptides, and the possibility of regulating the release of hydrophobic drugs by adjusting the hydrophobic amino acid side chain and the concentration of peptides.
Figure 15. Release profiles of EM from EM water suspension and in situ peptide-EM hydrogels (n = 3). [EM] = 1.0 mg/mL, [RADA16-I] = [RVDV16-I] = 0, 3, 5, 7 mg/mL (0 mg/mL represents EM water suspension without any peptide).
![Figure 15. Release profiles of EM from EM water suspension and in situ peptide-EM hydrogels (n = 3). [EM] = 1.0 mg/mL, [RADA16-I] = [RVDV16-I] = 0, 3, 5, 7 mg/mL (0 mg/mL represents EM water suspension without any peptide).](/cms/asset/dff5d958-c6b9-480c-842b-6b12d745af70/ianb_a_1673768_f0015_c.jpg)
Comparison of cellular viability of the two forms of EM
Though EM has been researched in many studies, its poor solubility, relatively difficult the route of administration and the inaccurate dosage made its pharmacological effects cannot be exerted effectively [Citation50,Citation51]. Due to high drug loading, simple preparation, diversified drug delivery routes and high drug dispersion [Citation52–55] nanosuspension is believed as a relatively simple and reliable method to improve the administration of insoluble drugs [Citation56]. In this study, colloidal suspensions of hydrophobic EM, which can form hydrogels in situ when they were introduced into physiological ions and/or pH conditions, were prepared with the self-assembling peptides RADA16-I and RVDV16-I. The cell viability of the peptide-EM in situ hydrogel on A549 and HepG2 cells was examined by MTT.
The results were shown in . With increased incubation time and dose increments, the anti-proliferative effect of EM and peptide-EM hydrogels on the A549/HepG2 cells both increased. The results indicated that cell viability in the group treated with RADA/RVDV-EM hydrogels were significantly lower than that in control, particularly after 48 h of incubation. These data suggested that RADA16-I-EM and RVDV16-I-EM hydrogel can maintain or improve EM’s inhibitory activity on tumor cells. It was concluded that the hydrogels formed by the self-assembling peptide RADA16-I or RVDV16-I can both effectively control the release of EM loaded in them, and prolong the inhibitory effect of EM on growth of A549 and HepG2 cells in vitro. This advantage for drug delivery systems will be explored in further in vivo experiments.
Figure 16. Proliferation inhibition effects of EM-water suspension and peptide-EM in situ hydrogel on A549 and HepG2 cells. [RADA16-I] = [RVDV16-I] = 5 mg/mL, [EM] = 20, 40, 60, 80, 100, 120,140, 160 μM; Data were calculated from three independent experiments. *p < .05, #p < .01. RADA: self-assembling RADA16-I peptide; RVDV: self-assembling RVDV16-I peptide; EM: emodin.
![Figure 16. Proliferation inhibition effects of EM-water suspension and peptide-EM in situ hydrogel on A549 and HepG2 cells. [RADA16-I] = [RVDV16-I] = 5 mg/mL, [EM] = 20, 40, 60, 80, 100, 120,140, 160 μM; Data were calculated from three independent experiments. *p < .05, #p < .01. RADA: self-assembling RADA16-I peptide; RVDV: self-assembling RVDV16-I peptide; EM: emodin.](/cms/asset/30ac3590-c34a-462a-95c1-f0a4e58f3885/ianb_a_1673768_f0016_b.jpg)
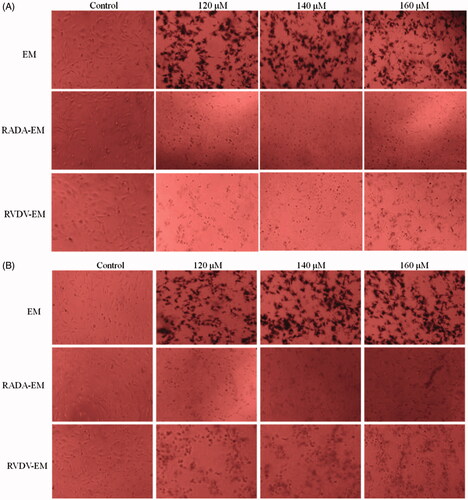
As shown in , after 48 h of treatment with the peptide-EM in situ hydrogel, the growth density and morphology of the cells changed. Under the microscope, the in situ hydrogel was observed covering the cell surface. The cell density in the hydrogel group was significantly lower than that in control; with the increase of EM concentration, a large amount of EM crystals can be clearly observed in the free EM group, while there was no obvious EM crystals can be seen in the hydrogel group. And the EM in the hydrogel inhibited the growth of tumor cells more significantly than the free EM. The in situ hydrogels formed self-assembling peptides-EM colloidal suspensions can improve the solubility of EM, increase the stability of EM in the mixture, control the release of EM and thus can help EM fully exert its anti-tumor effect.
Figure 17. A549 and HepG2 cells incubated with self-assembling peptide-EM hydrogel for 48 h. The morphological changes of A549 and HepG2 cells were observed using microscopy (magnification, ×200). A and B represent A549 and HepG2 cells, respectively. RADA: self-assembling RADA16-I peptide; RVDV: self-assembling RVDV16-I peptide; EM: emodin.
Conclusion
Both RADA16-I and RVDV16-I self-assembling peptides can interact with emodin in aqueous solution, suspension and hydrogels, and the interaction is particularly stronger for RVDV16-I peptide though the delivery performance of the two systems formed from the two different peptides have shown no difference. Both peptides can form colloidal suspension with emodin in aqueous solutions under magnetic stirring, from which in situ hydrogels under physiological conditions can be formed. In vitro release of emodin from the hydrogels showed a sustainably releasing phenomenon, which depends on the type of peptide and the concentrations. The formulation of suspension-in-situ hydrogel improved the administration of emodin, and strengthened the inhibition effects of free emodin on A549 and HepG2 cells in vitro. These results indicate that the self-assembling peptides have the potential as carriers of hydrophobic drugs like emodin. Both self-assembling peptides can allow hydrophobic drugs to be relatively stable in aqueous body fluid systems and released from in-situ hydrogels of self-assembling peptides, and the release rate can be adjusted by adjusting the type and concentration of the peptides. Thus the self-assembling peptides can be used to construct the suspension-in-situ hydrogel delivery system for hydrophobic drugs to improve the administration with maintaining or promoting effects. More interaction mechanisms and application details need to be studied by further in vivo researches.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Shi Y, Li J, Ren Y, et al. Pharmacokinetics and tissue distribution of emodin loaded nanoemulsion in rats. J Drug Deliv Sci Tec. 2015;30:242–249.
- Shrimali D, Shanmugam MK, Kumar AP, et al. Targeted abrogation of diverse signal transduction cascades by emodin for the treatment of inflammatory disorders and cancer. Cancer Lett. 2013;341(2):139–149.
- Ahn SM, Kim HN, Yu RK, et al. Emodin from polygonum multiflorum, ameliorates oxidative toxicity in HT22 cells and deficits in photothrombotic ischemia. J Ethnopharmacol. 2016;188:13–20.
- Huang J, Gong W, Chen Z, et al. Emodin self-emulsifying platform ameliorates the expression of FN, ICAM-1 and TGF-β1 in AGEs-induced glomerular mesangial cells by promoting absorption. Eur J Pharm Sci. 2017;99:128–136.
- Lu J, Ying X, Zhe Z, et al. Emodin suppresses proliferation, migration and invasion in ovarian cancer cells by down regulating ILK in vitro and in vivo. OTT. 2017;10:3579–3589.
- Iwanowycz S, Wang J, Hodge J, et al. Emodin inhibits breast cancer growth by blocking the tumor-promoting feedforward loop between cancer cells and macrophages. Mol Cancer Ther. 2016;15(8):1931–1942.
- Haque E, Kamil M, Irfan S, et al. Blocking mutation independent p53 aggregation by emodin modulates autophagic cell death pathway in lung cancer. Int J Biochem Cell B. 2018;96:90–95.
- Xie QC, Yang YP. Anti-proliferative of physcion 8-O-β-glucopyranoside isolated from Rumex japonicus Houtt. on A549 cell lines via inducing apoptosis and cell cycle arrest. BMC Complem Alter. 2014;14:1–10.
- Su J, Yan Y, Qu J, et al. Emodin induces apoptosis of lung cancer cells through ER stress and the TRIB3/NF-κB pathway. Oncol Rep. 2017;37(3):1565–1572.
- Xing J, Song G, Deng J, et al. Antitumor effects and mechanism of novel emodin rhamnoside derivatives against human cancer cells in vitro. PLoS One. 2015;10(12):e0144781.
- Zhang L, He D, Li K, et al. Emodin targets mitochondrial cyclophilin D to induce apoptosis in HepG2 cells. Biomed Pharmacoth. 2017; 90:222–228.
- Hsu CM, Hsu YY, Shieh FK, et al. Emodin inhibits the growth of hepatoma cells: finding the common anti-cancer pathway using Huh7, Hep3B, and HepG2 cells. Biochem Bioph Res. 2010;392(4):473–478.
- Hwang SY, Heo K, Kim JS, et al. Emodin attenuates radioresistance induced by hypoxia in HepG2 cells via the enhancement of PARP1 cleavage and inhibition of JMJD2B. Oncol Rep. 2015;33(4):1691–1698.
- Wang CG, Zhong L, Liu YL, et al. Emodin exerts an antiapoptotic effect on human chronic myelocytic leukemia K562 cell lines by targeting the PTEN/PI3K-AKT signaling pathway and deleting BCR-ABL. Integr Cancer Ther. 2017;16(4):526–539.
- Wang CG, Yang JQ, Liu BZ, et al. Anti-tumor activity of emodin against human chronic myelocytic leukemia K562 cell lines in vitro and in vivo. Eur J Pharmacol. 2010;627(1):33–41.
- Li BJ, Liu TB, Wang WF, et al. Effect of a novel emodin derivative on chronic myelogenous leukemia K562 cells and imatinib-resistant K562/G01 cells. J Exp Hematol. 2016;24:1–7.
- Dong H, Wu G, Xu H, et al. N-acetylaminogalactosyl-decorated biodegradable PLGA-TPGS copolymer nanoparticles containing emodin for the active targeting therapy of liver cancer. Artif Cells Nanomed Biotechnol. 2018;1:1–13.
- Liu H, Xu H, Zhang C, et al. Emodin-Loaded PLGA-TPGS nanoparticles combined with heparin sodium-loaded PLGA-TPGS nanoparticles to enhance chemotherapeutic efficacy against liver cancer. Pharm Res. 2016;33(11):2828–2843.
- Liu H, Gao M, Xu H, et al. A promising emodin-loaded poly (lactic-co-glycolic acid)-d-α-tocopheryl polyethylene glycol 1000 succinate nanoparticles for liver cancer therapy. Pharm Res. 2016;33(1):217–236.
- Morsi N, Ghorab D, Refai H, et al. Ketoroloac tromethamine loaded nanodispersion incorporated into thermosensitive in situ gel for prolonged ocular delivery. Int J Pharm. 2016;506(1–2):57–67.
- Chu K, Chen L, Xu W, et al. Preparation of a paeonol-containing temperature-sensitive in situ gel and its preliminary efficacy on allergic rhinitis. IJMS. 2013;14(3):6499–6515.
- Barron V, Killion JA, Pilkington L, et al. Development of chemically cross-linked hydrophilic-hydrophobic hydrogels for drug delivery applications. Eur Polym J. 2016;75:25–35.
- Liang J, Susan SX, Yang Z, et al. Anticancer drug camptothecin test in 3D hydrogel networks with HeLa cells. Sci Rep. 2017;7(1):37626.
- Raphael B, Khalil T, Workman VL, et al. 3D cell bioprinting of self-assembling peptide-based hydrogels. Mater Lett. 2017;190:103–106.
- Wu X, He L, Li W, et al. Functional self-assembling peptide nanofiber hydrogel for peripheral nerve regeneration. Regen Biomater. 2017;4(1):21–30.
- Xing R, Li S, Zhang N, et al. Self-assembled injectable peptide hydrogels capable of triggering antitumor immune response. Biomacromolecules. 2017;18(11):3514–3523.
- Wan S, Borland S, Richardson SM, et al. Self-assembling peptide hydrogel for intervertebral disc tissue engineering. Acta Biomater. 2016;46:29–40.
- Nune M, Krishnan UM, Sethuraman S. PLGA nanofibers blended with designer self-assembling peptides for peripheral neural regeneration. Mat Sci Eng C. 2016;62:329–337.
- McCloskey AP, Gilmore BF, Laverty G. Evolution of antimicrobial peptides to self-assembled peptides for biomaterial applications. Pathogens. 2014;3(4):791–821.
- Yang S, Wei S, Mao Y, et al. Novel hemostatic biomolecules based on elastin-like polypeptides and the self-assembling peptide RADA-16. Bmc Biotechnol. 2018;18(1):12–20.
- Acar H, Srivastava S, Chung EJ, et al. Self-assembling peptide-based building blocks in medical applications. Adv Drug Deliver Rev. 2017;110–111:65–79.
- Kuang H, Ku SH, Kokkoli E. The design of peptide-amphiphiles as functional ligands for liposomal anticancer drug and gene delivery. Adv Drug Deliver Rev. 2017;110–111:80–101.
- Griffin BT, Guo J, Presas E, et al. Pharmacokinetic, pharmacodynamic and biodistribution following oral administration of nanocarriers containing peptide and protein drugs. Adv Drug Deliver Rev. 2016;106:367–380.
- Koutsopoulos S. Self-assembling peptides in biomedicine and bioengineering: tissue engineering, regenerative medicine, drug delivery, and biotechnology. Peptide Appl Biomed Biotechnol Bioeng. 2018;15:387–408.
- Rymer SJ, Tendler SJ, Bosquillon C, et al. Self-assembling peptides and their potential applications in biomedicine. Ther Deliv. 2011;2(8):1043–1056.
- Eskandari S, Guerin T, Toth I, et al. Recent advances in self-assembled peptides: implications for targeted drug delivery and vaccine engineering. Adv Drug Deliver Rev. 2017;110-111:169–187.
- Ruan L, Zhang H, Luo H, et al. Designed amphiphilic peptide forms stable nanoweb, slowly releases encapsulated hydrophobic drug, and accelerates animal hemostasis. Proc Natl Acad Sci USA. 2009;106(13):5105–5110.
- Liu J, Zhang L, Yang Z, et al. Controlled release of paclitaxel from a self-assembling peptide hydrogel formed in situ and antitumor study in vitro. Int J Nanomed. 2011;6:2143–2153.
- Ashwanikumar N, Kumar NA, Saneesh Babu PS, et al. Self-assembling peptide nanofibers containing phenylalanine for the controlled release of 5-fluorouracil. IJN. 2016;11:5583–5594.
- Hu C, Huang P. Recent research progress in the determination of solid solubility. J Pharm Anal. 2010;22(4):761–766.
- Yuan L, Zhong S, Zhou R, et al. Determination of the equilibrium solubility and apparent oil-water partition coefficient of cantharidin by HPLC. Chin J New Drugs. 2017;26(10):1185–1188.
- Li F, Ding Z, Cao Q. Determination of dissociation constants of five rhubarb anthraquinone derivatives. Chin J Tradit Chin Med. 2007;32(2):166–168.
- Bin Y. Study of emodin liposome. Dissertation. 2009.
- National Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China. Beijing (China): China Medical Science and Technology Press; 2015.
- Wu D, Zhang S, Zhao Y, et al. The effects of motif net charge and amphiphilicity on the self-assembly of functionally designer RADA16-I peptides. Biomed Mater. 2018; 13(3):035011.
- Sun Y, Zhang Y, Tian L, et al. Self-assembly behaviors of molecular designer functional RADA16-I peptides: influence of motifs, pH, and assembly time. Biomed Mater. 2016; 12(1):015007.
- Shamsi F. Investigation of human cell response to covalently attached RADA16-I peptide on silicon surfaces. Colloid Surface B. 2016;145:470–478.
- Yu Z, Xu Q, Dong C, et al. Self-assembling peptide nanofibrous hydrogel as a versatile drug delivery platform. CPD. 2015;21(29):4342–4354.
- Hattori T, Ishii K, Tominaga T, et al. A fluorescence study on the local environment of hydrogels: double-network hydrogels having extraordinarily high mechanical strength and its constituent single-network hydrogels. Chem Phys. 2013;419:172–177.
- Xing YX, Li MH, Tao L, et al. Anti-cancer effects of emodin on HepG2 cells as revealed by 1H-NMR based metabolic profiling. J Proteome Res. 2018;17:1–41.
- Shia CS, Hou YC, Tsai SY, et al. Differences in pharmacokinetics and ex vivo antioxidant activity following intravenous and oral administrations of emodin to rats. J Pharm Sci. 2010;99(4):2185–2195.
- Shen C, Shen B, Liu X, et al. Nanosuspensions based gel as delivery system of nitrofurazone for enhanced dermal bioavailability. J Drug Deliv Sci Tec. 2018;43:1–11.
- Wang Y, Zheng Y, Zhang L, et al. Stability of nanosuspensions in drug delivery. J Control Release. 2013;172(3):1126–1141.
- Wang Y, Wang C, Jing Z, et al. A cost-effective method to prepare curcumin nanosuspensions with enhanced oral bioavailability. J Colloid Interface Sci. 2017;485:91–98.
- Wang L, Du J, Zhou Y, et al. Safety of nanosuspensions in drug delivery. Nanomed. 2017;13(2):455–469.
- Pawar SS, Dahifale BR, Nagargoje SP, et al. Nanosuspension technologies for delivery of drugs. Nanosci Nanotech Res. 2017;4:59–66.