
Abstract
The regeneration of the skin is vital to our wound healing and skin repair abilities. Adult epidermal stem cells (ESCs) have been shown to have the potential to renew old and dead skin cells, and ESCs have been implemented in stem cell-based therapies. GPR30 is a G protein-coupled membrane receptor for oestrogen, which has been shown to regulate cell proliferation and programmed cell death. Here, we examined the biological function of GPR30 in isolated adult murine ESCs. We show that GPR30 is fairly expressed in ESCs and is repressed upon ultraviolet B (UV-B) treatment in a dose-dependent manner. The activation of GPR30 by its agonist G1 ameliorates UV-B induced cellular oxidative stress and induction of IL-6 and IL-8. Furthermore, G1 protects against UV-B-induced cell death and improves the viability of ESCs. G1 also suppresses UV-B-induced HMGB-1 expression and protects the capacity of ESCs from the harm by UV-B radiation. Mechanistically, we show that co-treatment with G1 rescues UV-B-induced reduced Wnt1, cyclin D1 and β-catenin production, indicating the involvement of conical Wnt/β-catenin. Collectively, our data indicate that the activation of GPR30 has a protective role in ESCs, and GPR30 agonist G1-mediated ESC protection has potential implications in stem cell-based therapies for skin diseases.
Introduction
The skin is the largest organ in the human body. Skin cells form a tight barrier to protect our body from pathogen invasion and harmful UV light. The skin epidermis is the top visible layer of skin and is constantly being renewed by epidermal stem cells (ESCs) to replace damaged or dead cells, thus maintaining the balance in epidermal homeostasis. ESCs are one type of stem cells which have high intrinsic proliferative potential but a slow cycle rate in the epidermis [Citation1]. ESCs are largely responsible for skin regeneration and injury repair, and ESC-based stem cell therapies represent a new concept for the treatment of skin disorders and non-healing wounds [Citation2]. In healthy conditions, ESCs generate transit-amplifying daughter cells for the replenishment of skin tissue. The daughter cells eventually undergo terminal differentiation as the new keratinized layer [Citation3]. Stem cell-based therapy is a promising treatment for several different types of dermatological diseases, including wound healing, genodermatoses, burns, stable vitiligo and other chronic inflammatory diseases [Citation4,Citation5]. Recently, adult skin-derived ESCs have been shown to be fully functional, thereby providing a feasible source of ESCs. Preclinical studies have established the successful isolation of functional ESCs from adult murine skin, thus providing a powerful tool to study the biology of ESCs in vitro [Citation6,Citation7].
Oestrogen is the sex hormone which regulates the function of the female reproductive system. Oestrogens are well known to regulate many genes by binding to and activating nuclear oestrogen receptors (ERα and ERβ). It is now known that oestrogens can also activate membrane-bound oestrogen receptors, including GPR30 [Citation8]. GPR30 (also named GPER) is an orphan oestrogen receptor which only binds cell membranes but cannot directly regulate gene expression like ERα/β. Activation of GPR30 using its specific agonist G1 has been reported to participate in a diversity of biological activities. For example, administration of G1 in SD rats alleviated traumatic brain injury (TBI)-induced hippocampal neuronal apoptosis by promoting microglial polarization to the M2 phenotype and increasing the ratio of Arg1/iNOS [Citation9]. Another study reported that using G1 noticeably reduced the increase in infarction volume, improved the neurological deficit, and ameliorated neuronal injuries induced by middle cerebral artery occlusion (MCAO) in ovariectomized female mice [Citation10]. G1 treatment can also improve contractile function and reduce infarct size in isolated rodent hearts in response to ischemia/reperfusion through reducing mitochondria permeability transition pore opening [Citation11]. However, there has been little research on the physiological function of GPR30 and the possible usage of G1 in skin. Here, we investigated the expression of GPR30 and its activation in the functional regulation of murine ESCs in vitro.
Materials and methods
Isolation and culture of ESCs
ESCs were isolated from the skin on the backs of 8-week-old C57BL/6 mice. Briefly, the mouse skin was dissected and placed in HBSS and minced into small pieces. The tissues were then digested with 0.25% trypsin and vortexed several times to ensure adequate mixing. The digested cells were then spun to remove the supernatant, and the pellets were suspended and seeded into a six-well culture plate in DMEM/F12 medium containing 4 ng/ml bFGF (Thermo Fisher Scientific, Waltham, MA). To identify ESCs, isolated cells were detected using a flow cytometer (Thermo Fisher Scientific, Waltham, MA) with following antibodies: integrin β1 (ab179471, Abcam, Cambridge, UK), keratin 19 (ab191208, Abcam, Cambridge, UK) and keratin 10 (ab76318, Abcam, Cambridge, UK) using a method as previously reported [Citation12,Citation13]. The percentages of integrin β1- and KRT19-positive cells were greater than 85%, and the percentage of KRT10-positive cells was lesser than 10%. The ESCs were passaged every 3–5 days and used for low passage numbers. ESCs were stimulated with ultraviolet B (UV-B) (50 mJ/cm2) with or without 7.5 and 15 μM G1 [Citation14] for 24 h.
RT-PCR and real-time PCR
Total RNA from ESCs was extracted. A total of 2 µg RNA was reverse transcribed into cDNA using a Kit (Applied Biosystems, Foster City, CA, USA), and the real-time PCR experiments were performed with using a SYBR Green I master mix (Applied Biosystems, Foster City, CA, USA). Results were assessed using the 2–ΔΔCT method.
Western blot analysis
Total cell lysate of ESCs was prepared by lysing the washed cells using RIPA buffer with protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO). A total of 20–40 µg cell lysates was loaded into 4–12% precasted NuPAGE gel and run for 2 h to separate the protein mixture. The gel was then sandwiched with filter paper and transferred to a 0.2 μM PVDF membrane using a wet transfer apparatus with 100 V for 2 h. The blots were then blocked and incubated with corresponding antibodies. The immunoblots were developed by Pierce™ SuperSignal West Pico Plus (catalogue # 32132).
Dihydroethidium staining
A total of 5 × 105 ESCs were seeded into a six-well cell culture plate. After the necessary stimulation, cellular reactive oxygen species (ROS) production was detected by dihydroethidium (DHE) staining. Briefly, ESCs were washed and incubated with serum-free DMEM containing 5 µM DHE at 37 °C for 30 min without light. Cells were then washed three times with PBS by gently shaking. Fluorescent images were visualized using a fluorescence microscope. The fluorescence intensity in ESCs was analyzed with ImageJ software 7.0 (NIH, Bethesda, MD) to reflect cellular ROS [Citation15].
Cell viability
MTT assay was performed by plating ESCs into 96-well tissue culture plates [Citation16]. In general, 5000–10,000 ESCs per well were cultured for 48 h to reach full confluence and then treated with UV-B or G1. At the end-of-treatment time point, 5 μM MTT solution was added and incubated at 37 °C for 4 h. The reaction was stopped by adding 200 μl DMSO into the medium. OD value at 570 nm was recorded. The final normalized absorbance values are equal to signal absorbance minus background absorbance.
Enzyme-linked immunosorbent assay
We measured the secreted levels IL-6, IL-8 and HMGB-1 via enzyme-linked immunosorbent assay (ELISA). ESC culture media was collected and filtered to remove debris for ELISA experiments. The ELISA kits used were purchased from R&D Systems (Minneapolis, MN, USA). The data were read and recorded by a spectrometry. The relative expression of these proteins was normalized to total loading protein and presented as fold change.
Statistical analysis
Experimental results are expressed as mean ± SEM. Comparisons between different groups were performed by ANOVA test in SPSS (Version 20, Chicago, IL, USA), followed by Bonferroni’s test. p < .05 was considered a significant difference.
Results
GPR30 is expressed in ESCs
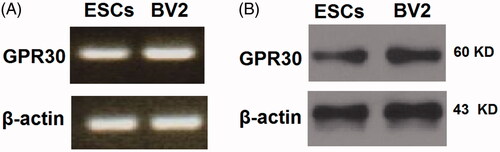
To investigate the role of GPR30 in ESCs, we first determined its expression level. In our experiment, we referenced the murine microglial BV2 cell line, which is known to express GPR30. The relative mRNA expression of GPR30 in ESCs is comparable to that in BV2 cells (. Via western blot analysis, we confirmed that GPR30 expression in ESCs is fair as referenced to BV2 cells (.
Figure 1. GPR30 is expressed in epidermal stem cells (ESCs). BV2 cells were used as a reference. (A) RT-PCR analysis of GPR30; (B) Western blot analysis of GPR30.
Ultraviolet B radiation reduces the expression of GPR30 in ESCs
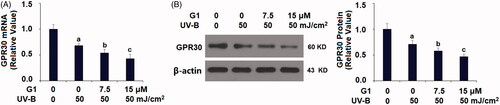
ESCs are sensitive to UV-B-induced cellular damage, and UV-B has been used as an effective approach to assess ESC function. We then challenged ESCs with UV-B radiation and examined its influence on GPR30 expression. In our experiment, we treated ESCs for 24 h with three different intensities: 25, 50 and 75 mJ/cm2. Our experiment showed that UV-B stimulation suppressed the mRNA of GPR30 in a dose-dependent manner (. We confirmed this dose-dependent suppression of UV-B on GPR30 protein expression (.
Figure 2. Ultraviolet B radiation reduces the expression of GPR30 in ESCs. ESCs were stimulated with ultraviolet-B (UV-B) with different intensities for 24 h. (A) mRNA of GPR30; (B) protein levels of GPR30 (a, b, c, p < .01 vs. previous group, n = 6).
GPR30 agonist G1 ameliorates UV-B radiation-induced oxidative stress
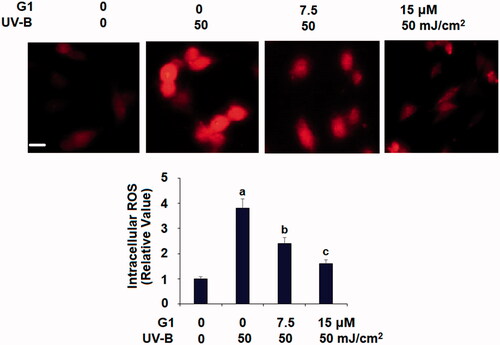
Based on the inhibition of GPR30 by UV-B, we hypothesized that the normal GPR30 level could be involved in the cellular effect of UV-B. We then co-treated ESCs with UV-B and the GPR30 inhibitor G1, and measured intracellular ROS levels. Compared with the non-treated control, UV-B induced roughly fourfold higher ROS production. However, co-treatment with UV-B and 7.5 or 15 μM G1 resulted in significantly lower ROS production, with the higher dose of G1 reducing ROS production to only 1.6-fold ().
Figure 3. Treatment with the specific GPR30 agonist G1 ameliorates UV-B radiation-induced oxidative stress in ESCs. ESCs were stimulated with ultraviolet-B (UV-B) (50 mJ/cm2) with or without 7.5, 15 μM G1 for 24 h. Intracellular ROS was determined by dihydroethidium (DHE) staining. Scale bar, 50 μm (a, b, c, p < .01 vs. previous group, n = 5–6).
GPR30 agonist G1 mitigates UV-B radiation-induced cytokine induction
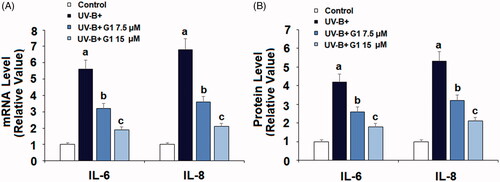
UV-B is known to induce cellular inflammation. Thus, we assessed the expression profile of IL-6 and IL-8. At the mRNA level and referenced to non-treated cells, UV-B induced 5–7-fold higher IL-6 and IL-8 mRNA expression, while co-treatment with the same two doses of G1 significantly lowered the transcription of these two cytokines, with the higher dose of G1 being more effective (. At the protein level, UV-B induced 4–5-fold higher IL-6 and IL-8 expression, but the presence of the same two doses of G1 significantly lowered their production, with the higher dose of G1 having a stronger inhibitory effect (.
Figure 4. Treatment with the specific GPR30 agonist G1 ameliorates UV-B radiation-induced expressions and secretions of pro-inflammatory cytokines. ESCs were stimulated with ultraviolet-B (UV-B) (50 mJ/cm2) with or without 7.5, 15 μM G1 for 24 h. (A) mRNA levels of IL-6 and IL-8; (B) ELISA analysis of IL-6 and IL-8 (a, b, c, p < .01 vs. previous group, n = 5–6).
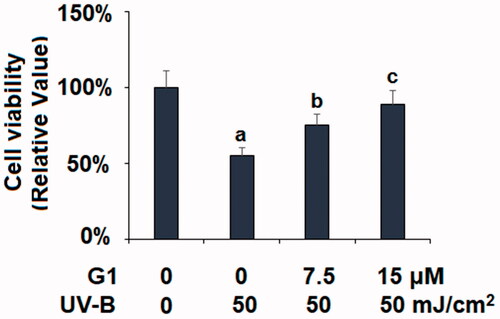
G1 mitigates UV-B-induced cell death
Next, we assessed the overall viability of ESCs in our co-treatment experiment via MTT assay. As compared with non-treated ESCs, the UV-B treatment group only had a 55% survival rate. But the two co-treatment groups (7.5 and 15 μM G1) showed improved survival rates (75 and 89%, respectively) (). This experiment showed that the GPR30 agonist G1 can protect ESCs from UV-B-induced cell death.
Figure 5. Treatment with the specific GPR30 agonist G1 restored UV-B radiation-induced reduction of cell viability. ESCs were stimulated with ultraviolet-B (UV-B) (50 mJ/cm2) with or without 7.5, 15 μM G1 for 24 h. Cell viability was determined by the MTT assay (a, b, c, p < .01 vs. previous group, n = 5–6).
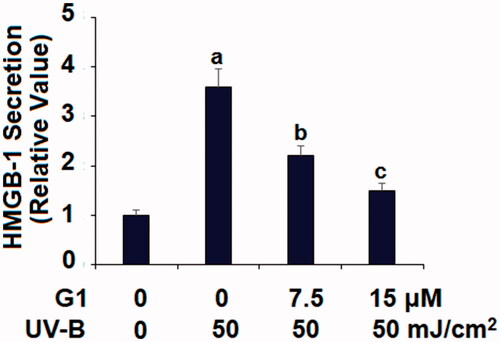
G1 suppresses UV-B-induced HMGB-1 production
HMGB-1 is a critical mediator of the late-stage inflammatory response, so we assessed its production in our co-treatment experiment. Referenced to non-treated cells, UV-B induced roughly fourfold higher HMGB-1 expression, but co-treatment with the two doses of G1 dramatically weakened its production, with the higher dose being more effective ().
Figure 6. Treatment with the specific GPR30 agonist G1 reduces UV-B-induced secretion of HMGB-1. ESCs were stimulated with ultraviolet-B (UV-B) (50 mJ/cm2) with or without 7.5, 15 μM G1 for 24 h. Secretion of HMGB-1 was determined by ELISA assay (a, b, c, p < .01 vs. previous group, n = 5–6).
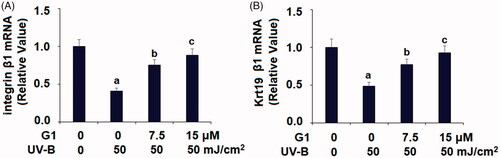
G1 protect ESCs from UV-B-induced loss of capacity
Next, we assessed the capacity of ESCs in our co-treatment experiment by examining the transcription of two signature factors – integrin β1 and the keratin protein Krt19. Compared to non-treated ESCs, UV-B treatment caused integrin β1 mRNA to be diminished by more than half. However, co-treatment with the two doses of G1 mitigated much the reduction in mRNA expression, with the higher dose having a better effect (. Similarly, UV-B treatment caused a reduction in Krt19 mRNA by about half. But co-treatment with the two doses of G1 attenuated the reduction in Krt19 induced by UV-B, with the higher dose being more effective (.
Figure 7. Treatment with the specific GPR30 agonist G1 protects ESCs against UVB-induced loss of capacities of ESCs. ESCs were stimulated with ultraviolet-B (UV-B) (50 mJ/cm2) with or without 7.5, 15 μM G1 for 24 h. (A) mRNA of integrin β1; (B) mRNA of Krt19 (a, b, c, p < .01 vs. previous group, n = 5–6).
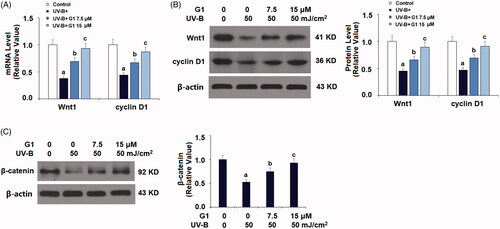
G1 rescues ESCs from UVB-induced inactivation of Wnt/β-catenin signalling
Finally, we explored the molecular pathways which could be involved in the activation of GPR30. We focussed on Wnt signalling due to its role in skin development. We tested several Wnt pathway genes, including Wnt1, cyclin D1 and its downstream target β-catenin. Compared to the non-treated control, both Wnt1 and cyclin D1 mRNA expression were reduced when ESCs were exposed to UV-B. But co-treatment with the two doses of G1 considerably ameliorated this reduction, with the higher dose more effectively (. We were able to confirm this effect at the Wnt1 and cyclin D protein levels (. We then checked the expression level of the molecule β-catenin, which is the downstream target of Wnt. UV-B treatment repressed β-catenin expression by about half, but co-treatment with the two doses of G1 almost rescued this repression, with the higher dose of G1 being more effective (.
Figure 8. Treatment with the specific GPR30 agonist G1 protects ESCs against UVB-induced inactivation of Wnt/β-catenin. ESCs were stimulated with ultraviolet-B (UV-B) (50 mJ/cm2) with or without 7.5, 15 μM G1 for 24 h. (A) mRNA of Wnt1 and cyclin D1; (B) protein expression of Wnt1 and cyclin D1; (C) protein expression of β-catenin (a, b, c, p < .01 vs. previous group, n = 5–6).
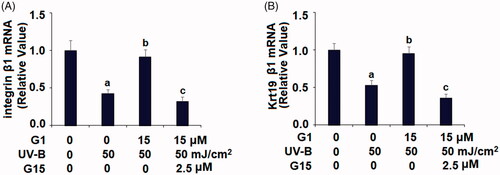
G15 abolishes the beneficial effects of G1 in maintaining the capacities of ESCs
To further confirm the role of GPR30 in G1-mediated protective effects against loss of the capacity of ESCs, G15, a selective antagonist of GPR30 was used. The real-time PCR results in indicate that treatment with G1 preserved UV-B-induced reduced integrin β1 and KRT19 expression. However, this beneficial effect of G1 was abolished when cells were co-stimulated with G15. These results suggest that G1 maintains the capacity of ESCs in a GPR30-dependent manner.
Figure 9. G15 abolished the protective effects of G1 in maintaining ESCs capacities. ESCs were stimulated with ultraviolet-B (UV-B) (50 mJ/cm2) with or without 15 μM G1 or 2.5 μM G15 for 24 h. (A) mRNA of integrin-β1; (B) mRNA of KRT19 (a, b, c, p < .01 vs. previous group, n = 6).
Discussion
As a membrane-bound orphan receptor for oestrogen, GPR30 has been shown to regulate second-message pathways and is an important regulator of cell growth and programmed cell death [Citation17]. Unlike ERα/β, GPR30 is primarily known as a mediator of rapid “nongenomic” effects. Its broad expression in different tissues has been shown to be involved in many aspects of reproduction, development, metabolic regulation and carcinogenesis, especially reproductive and breast cancers [Citation18]. GPR30 has specific binding activity for different oestrogen hormones. GPR30 activation by oestrogen or its agonist, G1 can promote tumour cell proliferation in female reproductive cancer cells [Citation18]. In the central nervous system, GPR30 and ERα/β are involved in oestrogen-mediated neuroprotection [Citation19]. In several different types of cancer cells, epidermal growth factor and TGF-α can transactivate the GPR30 promoter and promote its binding to ERα [Citation20]. This evidence suggests that GPR30 could act independently or interact with ERα/β to regulate critical cellular functions, including cell proliferation and survival. Previous research has revealed that the GPR30 agonist G1 activates ERK1/2 kinase [Citation21] and promotes cyclin D1 expression and proliferation in different cells [Citation22]. G1 also protects neurons from injury-induced apoptosis and microglial polarization [Citation9]. This evidence indicates that G1 compound could have a critical influence on cell function and possess protective roles in certain cell types.
In our study, we characterized the protective mechanism of GPR30 activation by its agonist G1 in ESCs. We isolated murine ESCs from adult animals and demonstrated that GPR30 is fairly expressed in ESCs. In cultured ESCs, we showed that GPR30 is a stress-responsive factor which is repressed by UV-B, suggesting that the natural expression level of GPR30 could be involved in the cellular response to UV-B treatment. Since the selective agonist of GPR30 G1 has been used as described before, we co-treated ESCs with G1 and UV-B, and assessed several aspects of the cellular response. It has been shown that the generation of ROS and inflammation induced by UV-B irradiation are the major contributors to cellular damage in ESCs [Citation23,Citation24]. We compared the consequences of UV-B irradiation with or without GPR30 activation by G1. The direct measurement of total cellular ROS indicated that activation of GPR30 ameliorated UV-B-associated ROS generation in ESCs to a great extent, while the production of IL-6 and IL-8 were dramatically reduced. These facts imply that the activation of GPR30 by G1 plays a protective role in ESC survival. Thus, we assessed the viability of ESCs in the presence or absence of G1 under UV-B treatment. Indeed, G1 co-treated ESCs showed a considerably higher survival rate than non-treated cells. Our data demonstrate that the anti-inflammatory and anti-ROS actions of GPR30 activation may be the pro-survival mechanism mediated by GPR30. We further explored the molecular pathways involving GPR30. Several signalling pathways have been shown to be closely associated with ESC renewal and skin regeneration. One of the major regulators of skin development is the Wnt pathway [Citation25,Citation26]. HMGB-1 protein is another important factor in late-stage inflammatory responses and skin tissue repair [Citation27]. We then explored their response to GPR30 activation. Our data demonstrate that G1 almost completely rescued UV-B-mediated Wnt pathway suppression as revealed by its rescue of Wnt1, cyclin D1 and β-catenin levels. Furthermore, we were able to confirm that G1 treatment also reduced UV-B-induced HMGB-1 induction. These data suggest that the conical Wnt1/β-catenin and HMGB-1 are the responsive pathways of GPR30 activation. Finally, we performed experiments to demonstrate that GPR30-mediated protection resulted in the preservation of the capacity of ESCs from UV-B irradiation. This was revealed by the ameliorative effect of G1 on UV-B triggered integrin-β and Krt19 reduction. Both integrin-β and Krt19 are the key signature molecules of epidermal derived stem cells [Citation3]. Therefore, the beneficial effect of G1 on ESCs preserved their capacity. We hypothesize that GPR30 activation mediates an unknown repairing mechanism to protect ESCs from exogenous irradiation.
In summary, our study demonstrates the protective mechanism of oestrogen receptor GPR30 signalling activation by its agonist G1 to resist UV-B irradiation-induced injury in ESCs. Our study revealed the unique role of GPR30 in response to UV-B irradiation to preserve the identity of ESCs, suggesting that the GPR30-mediated pathway is a pro-survival factor in ESCs. The protective mechanism of GPR30 activation illustrates another layer of regulation through which ESCs adapt to environmental harm. GPR30 could be an interesting target to optimize stem cell-based treatment of skin disorders.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Blanpain C, Fuchs E. Epidermal stem cells of the skin. Annu Rev Cell Dev Biol. 2006;22(1):339–373.
- Ojeh N, Pastar I, Tomic-Canic M, et al. Stem cells in skin regeneration, wound healing, and their clinical applications. Int J Mol Sci. 2015;16(10):25476–25501.
- Blanpain C, Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol. 2009;10(3):207–217.
- Prodinger CM, Reichelt J, Bauer JW, et al. Current and future perspectives of stem cell therapy in dermatology. Ann Dermatol. 2017;29(6):667–687.
- Jackson CJ, Tønseth KA, Utheim TP. Cultured epidermal stem cells in regenerative medicine. Stem Cell Res Ther. 2017;8(1):155.
- Moestrup KS, Andersen MS, Jensen KB. Isolation and in vitro characterization of epidermal stem cells. Methods Mol Biol. 2017;1553:67–83.
- Kostic L, Sedov E, Soteriou D, et al. Isolation of stem cells and progenitors from mouse epidermis. Curr Protoc Stem Cell Biol. 2017;41:1C.20.1–1C.20.11.
- Prossnitz ER, Arterburn JB, Sklar LA. GPR30: a G protein-coupled receptor for estrogen. Mol Cell Endocrinol. 2007;265–266:138–142.
- Pan MX, Tang JC, Liu R, et al. Effects of estrogen receptor GPR30 agonist G1 on neuronal apoptosis and microglia polarization in traumatic brain injury rats. Chin J Traumatol. 2018;21(4):224–228.
- Zhang Z, Qin P, Deng Y, et al. The novel estrogenic receptor GPR30 alleviates ischemic injury by inhibiting TLR4-mediated microglial inflammation. J Neuroinflammation. 2018;15(1):206.
- Bopassa JC, Eghbali M, Toro L, et al. A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2010;298(1):H16–H23.
- Zhang L, Ge Y, Fuchs E. miR-125b can enhance skin tumor initiation and promote malignant progression by repressing differentiation and prolonging cell survival. Genes Dev. 2014;28(22):2532–2546.
- Liu HW, Cheng B, Li JF, et al. Characterization of angiotensin-converting enzyme expression during epidermis morphogenesis in humans: a potential marker for epidermal stem cells. Br J Dermatol. 2009;160(2):250–258.
- Lin X, Li L, Wu S, et al. Activation of GPR30 promotes osteogenic differentiation of MC3T3-E1 cells: an implication in osteoporosis. IUBMB Life. 2019;71(11):1751.
- Cheng F, Yuan W, Cao M, et al. Cyclophilin A protects cardiomyocytes against hypoxia/reoxygenation-induced apoptosis via the AKT/Nox2 pathway. Oxid Med Cell Longev. 2019;2019:1.
- Sukovas A, Silkuniene G, Trumbeckaite S, et al. Hyperthermia potentiates cisplatin cytotoxicity and negative effects on mitochondrial functions in OVCAR-3 cells. J Bioenerg Biomembr. 2019;51(4):301.
- Feldman RD, Limbird LE. GPER (GPR30): a nongenomic receptor (GPCR) for steroid hormones with implications for cardiovascular disease and cancer. Annu Rev Pharmacol Toxicol. 2017;57(1):567–584.
- Filardo EJ, Thomas P. Minireview: G protein-coupled estrogen receptor-1, GPER-1: its mechanism of action and role in female reproductive cancer, renal and vascular physiology. Endocrinology. 2012;53(7):2953–2962.
- Liu SB, Zhao MG. Neuroprotective effect of estrogen: role of nonsynaptic NR2B-containing NMDA receptors. Brain Res Bull. 2013;93:27–31.
- Vivacqua A, Lappano R, De Marco P, et al. G protein-coupled receptor 30 expression is up-regulated by EGF and TGF alpha in estrogen receptor alpha-positive cancer cells. Mol Endocrinol. 2009;23(11):1815–1826.
- Gonzalez de Valdivia E, Broselid S, Kahn R, et al. G protein-coupled estrogen receptor 1 (GPER1)/GPR30 increases ERK1/2 activity through PDZ motif-dependent and -independent mechanisms. J Biol Chem. 2017;292(24):9932–9943.
- Sirianni R, Chimento A, Ruggiero C, et al. The novel estrogen receptor, G protein-coupled receptor 30, mediates the proliferative effects induced by 17beta-estradiol on mouse spermatogonial GC-1 cell line. Endocrinology. 2008;149(10):5043–5051.
- Panich U, Sittithumcharee G, Rathviboon N, et al. Ultraviolet radiation-induced skin aging: the role of DNA damage and oxidative stress in epidermal stem cell damage mediated skin aging. Stem Cells Int. 2016;2016:1.
- Doles J, Storer M, Cozzuto L, et al. Age-associated inflammation inhibits epidermal stem cell function. Genes Dev. 2012;26(19):2144–2153.
- Lim X, Tan SH, Koh WLC, et al. Interfollicular epidermal stem cells self-renew via autocrine Wnt signaling. Science. 2013;342(6163):1226–1230.
- Veltri A, Lang C, Lien WH. Concise review: Wnt signaling pathways in skin development and epidermal stem cells. Stem Cells. 2018;36(1):22–35.
- Straino S, Di Carlo A, Mangoni A, et al. High-mobility group box 1 protein in human and murine skin: involvement in wound healing. J Invest Dermatol. 2008;128(6):1545–1553.