
Abstract
The clinical benefit of cancer immunotherapy, including tumour vaccines, is influenced by immunosuppressive factors in the tumour microenvironment. Among these factors, cancer-associated fibroblasts (CAFs) and their products, such as fibroblast activation protein-α (FAPα), greatly affect tumourigenesis, development, metastasis and treatment tolerance, which make them promising immunotherapy targets for cancer patients. Our previous study reported that a whole cell tumour vaccine (WCTV) expressing FAPα inhibited tumour growth by simultaneously attacking cancer cells and CAFs. This study aimed to improve WCTVs with xenoantigens to end immune tolerance and to further activate the adaptive immune system. In the present study, we designed a WCTV by transducing a vector encoding human FAPα (hFAPα) into murine tumour cells and evaluated its efficacy in multiple solid tumour models. Immunotherapy with this WCTV effectively delayed tumour growth and prevented recurrence. The anti-tumour responses were clearly linked to antigen-specific cytotoxic T cells, whereas CD4(+) T lymphocytes also played a role. Humoural immune responses were activated because the adoptive transfer of immunoglobulins induced abscopal anti-tumour effects, and autoantibodies against FAPα were specifically detected in the sera of immunized mice. Moreover, an increased number of apoptotic tumour cells along with a reduced number of CAFs within the tumours suggest that xenogeneic FAPα-based WCTV has the potential to drive T cell and antibody responses against cancer cells and CAFs. This finding could offer an advanced strategy to treat multiple solid tumours with individualized cancer immunotherapy techniques.
Introduction
Although conventional therapies, such as surgery, radiation therapy and chemotherapy can cure cancer in approximately half of the cancer patients, in 2015, 2.814 million people died of the disease in China alone [Citation1]. In recent years, remarkable progress has been made in cancer immunotherapy, which stimulates the immune system to achieve a systemic and durable response to fight cancer, showing exciting long-term efficacy in people who developed the disease [Citation2]. Significant research efforts have focussed on whole cell tumour vaccines (WCTVs), which are produced with tumour cell lysates or inactivated whole tumour cells and can supply all relevant tumour-associated antigens (TAAs). In addition to having the potential to simultaneously activate CD8(+) cytotoxic lymphocytes (CTLs) and CD4(+) T helper cells [Citation3,Citation4], WCTVs are suitable for all patients regardless of their human lymphocyte antigen (HLA) type, which saves the trouble of defining tumour-derived proteins or peptides [Citation5].
However, the encouraging results of the early-phase studies are in marked contrast to the low clinical effectiveness observed in clinical studies [Citation6,Citation7]. This may be due to multiple factors, such as the tumour cells themselves having the characteristics of poor immunogenicity [Citation8,Citation9], the downregulated expression of major histocompatibility complex (MHC) [Citation10], the lack of co-stimulatory molecules, and immune suppression exerted by the tumour microenvironment (TME) [Citation11–13]. The TME consists of various cell types, extracellular matrices (ECMs), cytokines, chemotactic factors and mechanical cues that could expedite tumour transformation, harbour the tumour from host immunity, promote tumour growth and progression, reduce therapeutic effectiveness and provide niches for stagnant metastases to thrive [Citation14]. Accounting for a large proportion of cells in the TME, cancer-associated fibroblasts (CAFs) supply cancer cells with growth factors and ECM-degrading enzymes, remodel the ECM structure, and establish a local immunosuppressive microenvironment [Citation15,Citation16]. Fibroblast activation protein-α (FAPα), a transmembrane dipeptidyl peptidase (DPP), is expressed on the cell surface of CAFs at a high level across 90% of malignant tumour types. It affects various hormones and ECM components and is involved in all kinds of tumour-promoting activities [Citation17]. FAPα is virtually absent in normal tissues, except for in embryonic tissues, which makes it a selective and universal target [Citation18].
In recent years, the testing of diverse FAPα-targeted treatments has attracted significant research attention. Reisfeld et al. used an FAPα DNA vaccine to inhibit tumour cell growth in a murine breast cancer model, both alone and together with chemotherapeutic drugs [Citation19]. Loeffler et al. reported an oral vaccine targeting FAPα that can specifically kill CAFs and inhibit multi-drug-resistant tumour growth and metastases in murine breast cancer and colon cancer [Citation20]. Wen et al. used a cationic liposome-encapsulated FAPα plasmid as a vaccine to suppress tumour growth and metastases in a murine colon cancer model [Citation21]. Previously, we constructed a WCTV modified to express murine FAPα that can target both tumour cells and CAFs. Although the vaccination can slow tumour growth in the early stage, the tumour growth speeds up in the later stage [Citation22].
To make tumour vaccines more effective, xenogeneic homologous genes, which were identified in different species’ genome sequences, were used as antigens for cancer therapy [Citation23,Citation24]. For instance, Wei et al. reported that immunotherapy based on xenogeneic vascular endothelial cells induces an autoimmune-like reaction and destroys tumour angiogenesis, ultimately restraining tumour growth [Citation25]. He et al. showed that vaccination with xenogeneic homologous fibroblast growth factor receptor-1 (FGFR-1) produces autoantibodies against FGFR-1, which was linked to angiogenesis [Citation26]. Xenoantigens are seen as promising agents, not least because they differ from the body’s own protein composition, making their immunogenicity naturally superior to it. Moreover, although the heterologous antigens are mutated, they are still within the scope in which xenoantigens are still allowed to stimulate T cells via a cross-reaction [Citation27]. The homology of the gene sequence encoding FAPα in humans and mice is 90%; therefore, we tested the hypothesis that WCTV-expressing xenogeneic FAPα would enhance the vaccine’s immunogenicity, hinder immune effector inhibition, and potentially expand pre-existing anticancer immune responses. In this study, we developed a WCTV-expressing human FAPα (hFAPα) that could awaken the host adaptive immune systems via CD8+ T cell-mediated killing and specific autoantibodies against FAPα to inhibit tumour growth in multiple mouse solid tumour models.
Results
Tumour cells transfected with hFAP could express active hFAPα in vitro
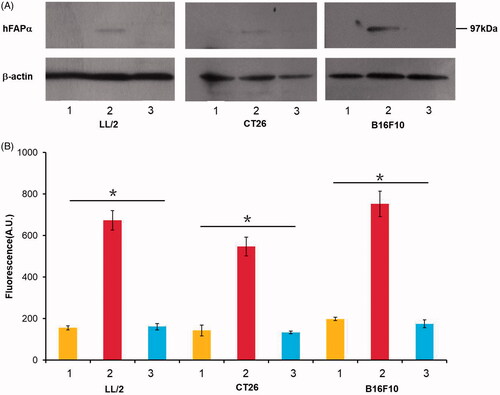
We first investigated whether murine tumour cells transfected with a hFAPα expression vector would express hFAPα in vitro. The protein level of hFAPα in tumour cells, including Lewis lung carcinoma LL/2, CT26 colon carcinoma and B16F10 melanoma, was measured by a Western blot. In tumour cells transduced with the hFAPα expression vector, a 97 kDa hFAPα was detected via a Western blot, while no binding site of hFAPα was found in wild-type tumour cells or cells transduced with an empty vector (). Furthermore, to demonstrate the biological activity of hFAPα produced by hFAP-transfected murine tumour cells in vitro, we then tested their DPP activity level. As shown in , the level of DPP activity was apparently higher in hFAP-transduced cancer cells than in other control groups (p < .05), which confirmed that hFAP-transfected murine tumour cells could express active hFAPα in vitro.
Figure 1. Tumour cells, including LL/2, CT26 and B16F10, were transfected with an hFAPα expression vector (line 2) and an empty vector pVector (line 1) via the Lipofectamine 3000 reagent, and wild-type tumour cells (line 3) were used as controls. (A) hFAPα expression levels in murine tumour cells analyzed by a Western blot. (B) The DPP activity levels of hFAPα produced by tumour cells. Data are expressed as the means ± SDs (n = 3), and one-way ANOVA was used to analyze the statistical significance. *p < .05. The DPP activity levels of hFAPα produced by tumour cells transfected with hFAPα expression vector compared to controls.
Therapeutic effect of a hFAPα-expressing WCTV
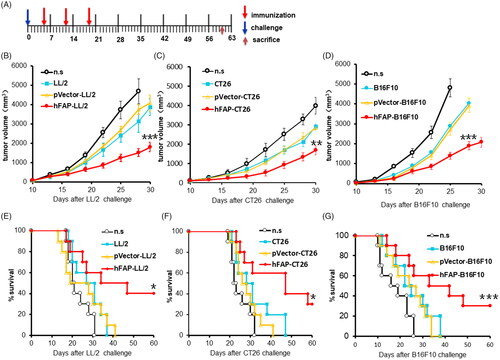
Having demonstrated the biological activity of hFAPα produced by hFAP-transfected murine tumour cells, we next attempted to test their immunotherapy effect in established tumour models, including Lewis lung carcinoma, CT26 colon carcinoma and B16F10 melanoma. Mice were vaccinated once a week for three weeks after the injection of cancer cells (). In the mouse LL/2 model, the subcutaneous (s.c.) administration of a traditional WCTV retarded tumour growth in some measurements due to poor immunogenicity, but once the mice were vaccinated with WCTV-expressing hFAPα, tumour growth was efficiently delayed (). Similar results were shown in the CT26 colon carcinoma model () and the B16F10 melanoma model (). In addition to the suppression of tumour growth, a significantly prolonged survival period also suggested a satisfactory immunotherapy effect of a WCTV expressing hFAPα on tumours ().
Figure 2. Vaccination with a hFAPα-expressing WCTV restrains the tumour growth of tumour-bearing mice. (A) After transplantation of 1 × 106 tumour cells into the right flank on day 0, mice were vaccinated with 1 × 106 inactivated hFAP-transduced, pVector-transduced cancer cells or wild-type cancer cells through subcutaneous injection (s.c.) on the bilateral axillaries and the unilateral inguinals on days 5, 12 and 19. Normal saline (n.s.) was used as a negative control. Each experimental group was tested twice. (B–D) The tumour volume in mice immunized with an hFAPα-expressing WCTV or controls in the three tumour models. Values are presented as the means ± SDs (n = 10). The statistical significance was analyzed using one-way ANOVA. **p < .01; ***p < .001 tumour volume of hFAP-LL/2, hFAP-CT26, hFAP-B16F10 differs from control groups. (E–G) Survival of mice after different vaccinations (n = 10). The log-rank (Mantel–Cox) test was utilized to calculate statistical significance. *p < .05; ***p < .001 survival of hFAP-LL/2, hFAP-CT26, hFAP-B16F10 compared to control groups.
Anti-tumour activity of a hFAPα-expressing WCTV involved cellular immune responses
To determine the potential role of cellular immune responses in the anti-tumour effect induced by a WCTV expressing hFAPα, we isolated T lymphocytes from vaccinated C57BL/6J mice and injected them into recipient mice intravenously (i.v.) via tail veins. The tumour growth in the mice from the group that received T lymphocytes from hFAP-LL/2 vaccinated mice was markedly decreased (), and the lifespans of the mice were correspondingly extended (). These findings suggested that cellular immune responses were involved in the magnified anti-tumour activity of immunotherapy with hFAP-LL/2 cells.
Figure 3. The anti-tumour effect of hFAP-LL/2 involved cellular immune responses. (A, B) Ability of the adoptive transfer of splenocytes from syngeneic mice to protect recipient mice from tumour growth in the LL/2 model. Splenocytes (1 × 107 cells per mouse) collected from different treatment groups were injected into the tail veins of the recipient mice. Significant differences in tumour volume of the mice that received spleen cells from hFAPα-expressing WCTV-treated mice differ from controls are presented by asterisks (***p < .001), and they exhibited a markedly increased survival rate compared to controls (***p < .001). (C) The revocation of the anti-tumour effect by the exhaustion of immunocyte subsets. The anti-tumour effect induced by hFAPα-expressing WCTVs was completely abrogated by the exhaustion of CD8+ T cells. Depletion of CD4(+) T cells partly affected the abrogation of anti-tumour activity, while the depletion of NK cells did not obstruct the development of anti-tumour immunity. (D) CTL-mediated cytotoxicity in vitro by the 51Cr release assay. T lymphocytes isolated from hFAPα-expressing WCTV-treated mice showed increased cytotoxicity to CAFs and LL/2 tumour cells (p < .05). (E) CD8 (FITC) and CD4 (PE) antibodies were used to stain single cell suspensions of the lymph nodes of nearby primary tumours harvested from immunized mice, and a remarkably increased percentage of CD8+ T cells was found in mice immunized with hFAP-LL/2.
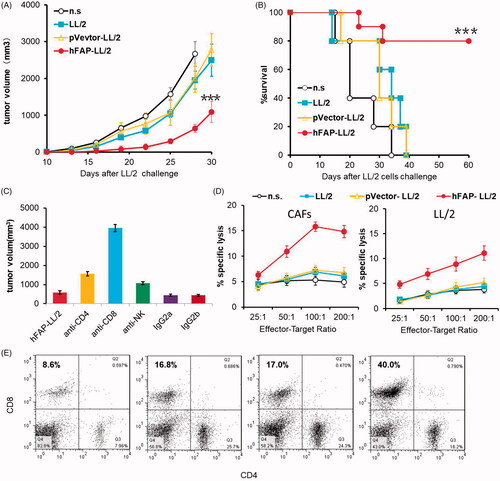
To explore which immune cell subsets play leading roles in the anti-tumour immune responses elicited by a WCTV expressing hFAPα, we administered corresponding monoclonal antibodies to deplete the mice of CD4(+) or CD8(+) T cells or natural killer (NK) cells in vivo. Cancer cells were injected s.c. into the right flanks of mice seven days after the last vaccination. As shown in , mice in the CD8(+) T lymphocyte depletion group in combination with hFAP-LL/2 cell vaccination were not protected from tumour growth, and we found that the CD4(+) T cell depletion before vaccination was, to some extent, linked to the abrogation of anti-tumour effects. In contrast, pre-treatment with monoclonal antibodies against NK cells had no effect on the development of anti-tumour immunity elicited by hFAP-LL/2 cell vaccination. These findings indicated that CD8(+) T cells may play a large role in anti-tumour activity and that CD4(+) T lymphocytes also played a role to a certain extent.
Importantly, we found that T cells obtained from mice treated with hFAP-LL/2 showed higher cytotoxicity against target cells, including LL/2 cancer cells and CAFs, especially the FAP-positive target cells, but T cells from mice immunized with pVector-LL/2, LL/2 or normal saline (n.s.) did not show increased cytotoxicity to LL/2 cells or CAFs (). This finding was in accordance with the CTL activity requirement for the anti-tumour effects induced by a hFAPα-expressing WCTV. Then, we used flow cytometry to identify effector T cells in the lymph nodes of nearby primary tumours to investigate the effect of vaccination on immune effector cells near the tumour. As shown in , vaccination with hFAP-LL/2 obviously increased the relative percentage of CD8(+) T lymphocytes in the lymph nodes of nearby primary tumours compared to other controls, indicating that immunization with a hFAPα-expressing WCTV effectively enhanced the recruitment of immune effector cells.
hFAPα-expressing WCTV decreases FAP expression and increases lymphocyte infiltration in tumour tissue
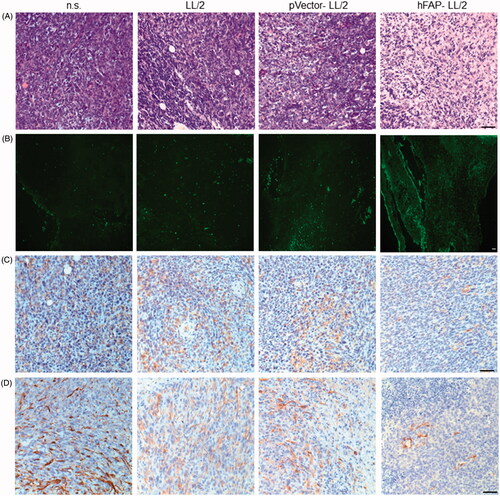
Having found that CD8(+) T cells are essential for the anti-tumour effects induced by hFAPα-expressing WCTVs, we next assessed whether lymphocyte infiltration was increased in the tumour tissues of immunized animals. As demonstrated via haematoxylin and eosin (H&E) staining, vaccination with hFAP-LL/2 cells was associated with increased eosinophil, monocyte and lymphocyte infiltration in tumour tissue, whereas only a few lymphocytes were detected in the tumours of mice treated with the control (). To investigate whether anti-tumour immunity is linked to the apoptosis of tumour cells, we carried out a terminal deoxynucleotidyl transferase-mediated nick end labelling (TUNEL) assay on tumour samples from immunized groups. The results showed that apoptotic cells in the tumours of mice treated with hFAP-LL/2 increased compared with those in the other control groups (). To determine whether the vaccine also targeted CAFs, the expression levels of two biomarkers of CAFs, FAPα and alpha smooth muscle actin (α-SMA), were initially evaluated using immunohistochemistry. As shown in , the expression levels of FAPα and α-SMA were higher in tumours from mice immunized with pVector-LL/2, LL/2 or n.s. than in those from hFAP-LL/2 vaccinated mice. An increased number of apoptotic tumour cells along with reduced CAFs within the tumours suggested that a WCTV based on xenogeneic FAPα has the potential to simultaneously target cancer cells and CAFs.
Figure 4. LL/2 tumour sections were harvested from C57BL/6J mice that were immunized once a week for three weeks after the tumour cell injection. (A, B) H&E staining and the TUNEL assay of tumour tissues. Tumour sections from mice treated with hFAP-LL/2 showed more lymphocytes and apoptotic cancer cells than those in the control groups. Scale bar, 100 µm. (C, D) Expression of FAPα and α-SMA within tumours from mice immunized with hFAP-LL/2, pVector-LL/2, LL/2 and normal saline. Scale bar, 100 µm.
Protective effect and humoural immune response evaluation
Vaccinated mice were challenged with cancer cells in line with the protocol described in the Methods section and . All mice treated with n.s. displayed very developed tumours, and tumour growth was partly suppressed in all mice vaccinated with irradiated pVector-transduced cancer cells or wild-type cancer cells. Once immunized with a WCTV-expressing hFAPα, three mice were found to be tumour-free in the LL/2 model, two mice were tumour-free in the B16F10 model, and the others were observed to have a remarkably slow rate of tumour growth (). These tumour-free mice were observed for more than 10 months, and no late tumour progression was found (data not shown). Effective protection against tumour growth could be very important for preventing tumour recurrence.
Figure 5. Protective anti-tumour responses. (A) C57 mice (n = 10) were vaccinated with irradiated hFAP-transduced, pVector-transduced, wild-type tumour cells or n.s. on D-21, D-14 and D-7. Mice were then injected subcutaneously with 1 × 106 tumour cells on D0. (B, C) There was a notable difference in tumour volume among vaccinated mice and those in the control groups. ***p < .001 tumour volume of hFAP-LL/2, hFAP-B16F10 differs from control groups. (D, E) Significant reduction in tumour-free rate. **p < .01; ***p < .001 tumour free rate of hFAP-LL/2, hFAP-B16F10 compared to controls.
The adoptive transfer of purified immunoglobulins obtained from hFAP-LL/2-immunized animals greatly reduced the tumour growth as well as remarkably prolonged the survival of the animals compared to those in the control groups (). To further investigate the mechanism of this specific immune response, we stained hFAP-LL/2 cancer cells, pVector-LL/2 cancer cells and untreated LL/2 cancer cells with sera obtained from hFAP-LL/2-vaccinated mice or control groups, and the stained cells were measured by flow cytometry to detect autoantibodies against FAPα. Polyclonal rabbit anti-FAPα antibody incubation cells were used as a positive control. In addition, hFAP-LL/2 cells exhibited obvious positive IgG staining with sera from hFAP-LL/2-immunized mice; this is in contrast to the negative staining for sera obtained from the control groups, as shown in . Conversely, pVector-LL/2 cells and untreated LL/2 cancer cells showed negative staining for immunized sera or control sera. All these findings indicate that the augmented anti-tumour effects of hFAP-LL/2 cells also involve humoural immune responses.
Figure 6. Anti-tumour activity of the hFAPα-expressing WCTV involved in humoural immune responses. (A, B) Ability of adoptive transfer of immunoglobulins from syngeneic mice to protect recipient mice from tumour growth in the LL/2 model. Purified immunoglobulins (300 µg/mg) from mice vaccinated with different vaccines were injected into the tail veins of recipient animals. Inhibition of subcutaneous tumour growth and apparently increased survival (***p < .001) was found when mice received immunoglobulins from hFAPα-expressing WCTV-treated mice. (C) Exponentially growing LL/2, pVector-LL/2 and hFAP-LL/2 tumour cells were incubated with purified immunoglobulins in mice from different vaccine groups. The polyclonal rabbit anti-FAPα antibody incubation cells were used as a positive control. Autoantibodies against FAPα were detected in the sera of hFAPα-expressing WCTV-immunized mice via flow cytometry.
General safety evaluation
The vaccinated mice were observed for possible side effects for a long-term (>10 months). We also evaluated the safety of as-designed WCTV. When mice were vaccinated with the WCTV, the bodyweight of hFAP-LL/2 immunized mice did not significantly fluctuate (Supplementary Figure S1). No adverse effects were found based on other gross measures of health such ruffling of fur, behaviour, life span or feeding.
Discussion
In this study, we constructed a WCTV by transducing a vector encoding hFAPα into murine tumour cells and evaluated its efficacy in multiple murine tumour models. The vaccine showed effective tumour growth delay and recurrence prevention in vivo. No obvious side effects were detected following vaccination. The adoptive transfer of T cells and the depletion of T lymphocytes revealed that CD8(+) T cells play a large part in the anti-tumour immune response, and CD4(+) T lymphocytes were also shown to play a role. Humoural immunity was also elicited because the adoptive transfer of the immunoglobulins induced a strong anti-tumour effect as well, and autoantibodies against FAPα were specifically detected in the sera of immunized mice. Moreover, the number of apoptotic tumour cells increased and CAFs apparently reduced within the tumours. These findings suggested that a WCTV based on xenogeneic FAPα may enhance not only antibody but also T cell responses to fight cancer cells and CAFs.
For decades, many immunotherapy strategies have been established to stimulate the patients’ own immune systems and to induce potent anti-tumour responses [Citation28]. Although various approaches based on tumour vaccines have been found to remarkably increase both innate and adaptive immune cells in tumours, most patients who received the vaccination eventually experienced tumour progression or recurrence [Citation8,Citation29]. Some studies suggest that the immunosuppressive effects of stromal cells that live nearby tumour cells are partly to blame. The successful development and final metastasis of tumours not only depend on the genetic changes of neoplastic cells but also involve the complex tissue environment. The TME is a complex integrated system in which immunosuppressive and immunosupportive mechanisms coexist, but the immunosuppressive mechanisms usually have the upper hand [Citation2], thus helping tumour cells resist the immune system attack:
In addition to the increased expression of immunosuppressive molecules (e.g. the Fas ligand) and increased cytokine (e.g. TGF-β and interleukin-10) delivery [Citation30,Citation31], cancerous cells can inhibit antigen presentation by selectively losing tumour-specific antigens and downregulating MHC-I molecule expression, thus directly avoiding T cell recognition [Citation32,Citation33] or by upregulating ligands, such as PD-L1, that inhibit T cell receptors, which leads to the transient activation or depletion of limited antigen-specific T cell populations and attenuates specific cytotoxicity during the immune response [Citation10,Citation11,Citation34];
The infiltration of various cell subpopulations in tumours blocks the cytotoxicity of CTL or NK/T cells to abnormal cells [Citation35];
A variety of cells in the TME form a coating on the tumour tissue, preventing immune cells from infiltrating the tumour site, reducing the entry of antineoplastic drugs and modulating the response of cancer cells to immunotherapy, chemotherapy or other treatments [Citation36].
Therefore, changing the cancer microenvironment is on track to become a new strategy for tumour immunotherapy, and much research has focussed on therapeutic protocols against TME elements, such as combined therapies and nanomedicines [Citation37–39]. Previous studies have shown that immunotherapy for cancer micrometastases employing an IL-21-based cellular vaccine can be significantly augmented through CD25+ regulatory T cell depletion [Citation40], and a combination of vaccination with agents that disarm TGFβ would enhance the vaccine’s efficacy [Citation41]. Unlike tumour cells, which are known to be genetically unstable, can accumulate adaptive mutations and eventually cause drug resistance, stromal cell types within the TME are more stable and are appropriate therapeutic targets for immunotherapy, reducing the incidence of drug resistance and tumour recurrence [Citation42]. Moreover, the downregulation of MHC-I molecule expression does not affect the antigen presentation process between stromal fibroblasts and the TCR complex [Citation28]. Furthermore, CAFs can be against in vivo or in situ, or they can be manipulated in vitro and then reinjected back into the same patient [Citation17]. Thus, targeting CAFs could be an effective treatment choice for patients with different solid tumour types [Citation43]. In a previous study, we constructed a WCTV modified to express FAPα that elicited anti-tumour immunity against both cancer cells and CAFs mediated by CD8+ T cells, but the WCTV requires further improvement to increase its therapeutic effect [Citation22].
Most immunogenic tumour proteins are “self” proteins, and therefore, peripheral tolerance plays an important role in tumour immune escape [Citation41]. On this basis, it has been proposed that the natural immune tolerance of TAAs could be overcome by using homologous antigens from different species. These xenogeneic antigens would need to be sufficiently different from the autoantigens to make them more immunogenic, but at the same time, these antigens must be within the optimal range of homology that still allows them to induce cross-reactive T cells [Citation27]. Prior work has documented the potential of xenogeneic homologous genes to act as tumour antigens, eliminating immune tolerance for cancer immunotherapy. For example, Alexander et al. [Citation44] evaluated the efficacy of an allogeneic cancer cell vaccine enhanced by xenogeneic melanoma antigen in a melanoma model of pet dogs, and the vaccine resulted in an overall response rate of 17% and a cancer control rate of 35%. An early study showed that a DNA vaccine targeting hFAPα suppressed breast cancer growth by eliciting specific cellular immune responses and altering the TME in a mouse model [Citation45]. In the present study, we modified murine tumour cells expressing hFAPα as a WCTV to break the mouse immune tolerance to FAPα. This vaccine targets tumour cells and interstitial-based CAFs and augments cellular and humoural immune responses simultaneously in the most efficient manner.
In other words, the improvements found in this research are related not only to cellular immune responses but also to humoural immune responses because autoantibodies against FAPα were specifically detected in the sera of immunized mice. However, the host’s immune system does not normally produce autoantibodies against FAPα. The immunization process elicited by inactivated tumour cells expressing xenogeneic FAPα may impel the production of antibodies that cross-react with the intact FAPα expressed on the CAFs of the host. A similar pattern of results was obtained in a previous study that showed that whole tumour cells transfected with xenogeneic matrix metalloproteinase-2, a type IV collagenase expressed in tumour cells, induced autoreactive humoural and cellular immune responses as an alternative DNA vaccine [Citation46]. The present research confirms and extends previous findings, offering the possibility of using a xenogeneic stroma-antigen-enhanced tumour cell vaccine to simultaneously target cancer and immunosuppressive stromal cells.
There are limitations to the current study. We must point out that we focus on only the design and efficacy of the xenogeneic FAPα-based WCTV, its safety has not been well described, and future study will concentrate on more systematic safety evaluation such as hepatic, renal and cardiac toxicity. In addition, although our hypothesis was supported statistically, future research on the function of CAFs and the immune-related changes within the TME might extend the explanation of anti-tumour immunity elicited by this vaccine.
In conclusion, we developed a WCTV-expressing hFAPα and evaluated its efficacy in multiple murine tumour models. The vaccine showed satisfactory effects on tumour growth delay and relapse prevention. Further investigations indicate that the xenogeneic FAPα-based WCTV could awaken the host’s adaptive immune system and simultaneously enhance the antibody and T cell responses to fight cancer cells and CAFs. These findings suggest that xenogeneic FAPα-based WCTVs could elicit a strong therapeutic anti-tumour immune response and may serve as a potent agent for cancer immunotherapy.
Methods
Cells and mice
The mouse Lewis lung cancer cell line LL/2, colon cancer cell line CT26 and melanoma cell line B16F10 were ordered from ATCC (Manassas, VA). B16F10 and LL/2 cells were grown in Dulbecco’s modified Eagle medium (DMEM), and CT26 cells were grown in RPMI 1640 medium, each of which was supplemented with 10% (v/v) foetal bovine serum (Invitrogen, Carlsbad, CA, USA), penicillin and streptomycin in an incubator at 37 °C in 5% CO2. Female C57BL/6J and BALB/c mice (6–10 weeks) were ordered from the Laboratory Animal Center of Sichuan University (Chengdu, China). We chose all female mice in the study because they are not only more responsive and immune sensitive than males but also easy to maintain in a group. All animal experiments were conducted in accordance with protocols approved by the Animal Care and Use Committee of Sichuan Cancer Hospital.
Vaccine preparation
The coding sequences of hFAPα ordered from Open Biosystems (Huntsville, AL) were amplified and sub-cloned into the EcoRI and XhoI sites of PVITRO2-neo-mcs (pVector; InvivoGen, San Diego, CA, USA) to generate the hFAPα expression vector (hFAP), as described previously [Citation21]. DNA for transfections was produced by EndoFree Kits (Qiagen, Valencia, CA, USA), and each resulting construct was confirmed by sequencing. Tumour cells were transiently transduced with a reconstructed plasmid or with an empty vector via Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer’s instructions. After transfection and washing three times with phosphate-buffered saline (PBS), cancer cells were then inactivated by radiation (100 Gy) for in vivo use.
Western blot analysis
Thirty micrograms of protein (measured by a bicinchoninic acid protein assay kit, BCA) was separated on a 10% SDS-PAGE gel and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA). After blocking the non-specific binding, we incubated the blots with specific antibodies against FAPα (1:1000; Abcam, Cambridge, UK) or β-actin (Abcam, Cambridge, UK). Then, a horseradish peroxidase-conjugated goat anti-rabbit IgG (Abcam, Cambridge, UK) was incubated as the secondary antibody. Finally, we used enhanced chemiluminescence detection (Perkin-Elmer, Waltham, MA) to visualize the protein bands.
DPP activity
We used Ala-Pro-AFC (Bachem, Torrance, CA) as a substrate to investigate the DPP activity of the proteins. We coated Fluoronunc MaxiSorb 96-well plates with antibodies against FAPα (100 μg/mL), washed them with PBS containing 0.1% Tween 20 (PBST), and blocked them with 5% BSA [Citation47]. The protein concentration from tumour cells was measured, one microgram of protein was added to each well after incubation for 1 h, and the samples were washed with PBST. Finally, we evaluated the DPP activity by Ala-Pro-AFC (0.25 mmol/L) cleavage for one hour at room temperature. To quantitatively measure DPP activity, the release of free Ala-Pro-AFC was assessed in a Cytofluor fluorometer (Labsystem, Helsinki, Finland) at settings of 395/490 nm (excitation/emission).
Therapeutic study
To explore the therapeutic effects, mice were injected s.c. with cancer cells (1 × 106 cells/100 μL PBS/mouse) at the right flank. Five days later, they were randomized into four groups (n = 10) and treated with 1 × 106 inactivated hFAP-transduced, pVector-transduced cancer cells or wild-type cancer cells injected s.c. on the bilateral axillaries and the unilateral inguinal on days 5, 12 and 19. Then, n.s. was used as a negative control. The tumour size was measured with Vernier callipers, and then the tumour volume was determined by the following equation: width2 × length × 0.52 [Citation48]. Each experimental group was tested twice.
Adoptive transfer in vivo
We used 1 × 106 inactivated hFAP-transduced, pVector-transduced, non-transduced cancer cells and n.s. to vaccinate C57BL/6J mice on days 28, 14 and 7. Lymphocytes were isolated, and pooled serum was obtained from the vaccinated or control mice on day 0; the recipient mice were injected with freshly isolated T cells (1 × 107) through the tail vein one day after the LL/2 cell challenge. Another group of recipient mice was injected i.v. with immunoglobulin purified from pooled serum at 50 μg per kilogram one day before implantation with tumour cells. After that, immunoglobulin transfer was administered every three days for three weeks. Tumour growth was monitored, and mouse survival was recorded.
Immune cell subset depletion in vivo
We exhausted immune cell subsets according to previously published methods [Citation21]. In short, one day before the immunization, the animals were administered intraperitoneally (i.p.) with anti-mouse CD8 (clone 2.43, rat IgG), anti-mouse CD4 (clone GK 1.5, rat IgG), anti-NK (clone PK136) monoclonal antibodies or an isotype control at a dose of 500 µg/mouse. After that, we carried out this procedure two times per week for three weeks. The mice were injected with LL/2 cells seven days after the third vaccination. The tumour volumes were recorded 25 days after implantation.
CAF isolation and culture
CAFs were isolated from the tumour tissues of LL/2 tumour-bearing mice using the out-growth method and were identified using specific antibodies. In brief, the tissue was cut into finely divided pieces in a tissue culture dish using a scalpel, digested with 1 mg/mL type IV collagenase (Sigma-Aldrich, St. Louis, MO, USA) for 1 h at 37 °C and shaken frequently. Pure fibroblast cultures were obtained by cells selected from the primary culture mix and by supplementary cell propagation with 10% FBS. Cells were cultured under 3% O2 conditions and collected for a cytotoxicity assay after the second passage (two to three weeks). Cell authentication and purity were determined by the flow cytometry analysis of cells exposed to αSMA and FAPα.
In vitro target cell cytotoxicity assay
The cytotoxic activity of the vaccine was investigated using the 51Cr release assay as described by others and by us previously [Citation21,Citation22]. Effector cells (splenocytes isolated from mice) were seeded into 96-well plates in 100 μL aliquots/well. LL/2 cells and CAFs were used as target cells. 51Cr-labelled target cells were added to the effector cells at the indicated ratio of effector cells to the target (E:T). They were incubated for 18 h at 37 °C in CO2 incubators. The percentage cytotoxicity was calculated based on the following formula: (test 51Cr release–spontaneous 51Cr release)/(maximum 51Cr release–spontaneous 51Cr release)×100.
Flow cytometry
The lymph nodes of nearby tumours were chopped into single cell suspensions. Cells were washed twice with PBS. Cells were resuspended in binding buffer at 1 × 105 cells/100 μL and were added to FITC-conjugated CD8a (BD Biosciences, San Jose, CA) and to PE-conjugated CD4 (BD Biosciences, San Jose, CA, USA) to detect effector T cells [Citation49]. To detect FAPα-specific neutralizing antibodies in sera, 3 × 105 hFAP-transduced, pVector-transduced or wild-type B16F10 cells were seeded into a six-well plate and then cultured overnight in a CO2 incubator. The next day, cells were washed with PBS, fixed in 4% paraformaldehyde for 30 min, and incubated with 0.5% Triton X-100 for 20 min at room temperature, followed by 10 washes with PBS. Then, they were blocked with PBS + 2% BSA for 20 min at 4 °C and incubated with sera (diluted 1:100 by PBS and 2% BSA) from mice of each group for 1 h at 4°C. Next, they were incubated with fluorescently labelled anti-mouse IgG antibodies for 30 min at 4°C after three washes with PBS and 1% BSA. The cells incubated with the polyclonal rabbit anti-mouse FAPα antibody were used as a positive control. The samples were analysed by a BD FACSCalibur flow cytometer.
Immunohistochemistry staining
The formalin-fixed, paraffin-embedded tumour sections were deparaffinized, rehydrated and finally submerged in PBS. Histological sections of tumours were analysed via H&E staining, as described previously. After blocking endogenous peroxidase and the retrieval of the antigens, the slides were incubated at 37 °C with rabbit anti-mouse FAPα (1:100) and rabbit anti-mouse αSMA (1:1000), and the anti-sheep HRP-DAB Staining Kit (R&D Systems, Minneapolis, MN) was then used. Finally, the sections were incubated with haematoxylin and imaged. The apoptosis of LL/2 tumour cells was analysed by an in situ TUNEL assay according to the manufacturer’s instructions (Promega, Madison, WI). The slides were examined by fluorescence microscopy.
Protective effect evaluation
To investigate the protective effects of a WCTV expressing hFAPα, mice were vaccinated with different immunogens, including irradiated hFAP-transduced, pVector-transduced and non-transduced tumour cells on days 28, 14 and 7, with the n.s. group acting as the control. Seven days after the last vaccination, the mice were injected s.c. with 1 × 106 tumour cells in the right flank.
General safety evaluation
To investigate the potential toxicity of WCTV expressing human FAPα, the bodyweight of each mouse was measured every day during treatment. Besides, the mice were continuously observed for gross measures of health, such as ruffling of fur, life span, behaviour and feeding.
Statistical analysis
The statistical significance of differences was determined by one-way ANOVA tests with Tukey post hoc analysis, two-way ANOVA tests with Bonferroni’s post hoc analysis or log-rank tests. Data are expressed as the means ± SDs when normally distributed. The findings were regarded as significant at p < .05 (*), p < .01 (**) and p < .001 (***).
Supplemental Material
Download ()Disclosure statement
No potential conflict of interest was reported by the authors.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
Related Research Data
References
- Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA: Cancer J Clin. 2016;66:115–132.
- Tang H, Qiao J, Fu YX. Immunotherapy and tumor microenvironment. Cancer Lett. 2016;370(1):85–90.
- Weiss EM, Wunderlich R, Ebel N, et al. Selected anti-tumor vaccines merit a place in multimodal tumor therapies. Front Oncol. 2012;2:132.
- Chiang CL, Benencia F, Coukos G. Whole tumor antigen vaccines. Semin Immunol. 2010;22(3):132–143.
- Chiang CL, Coukos G, Kandalaft LE. Whole tumor antigen vaccines: where are we? Vaccines. 2015;3(2):344–372.
- Itoh K, Yamada A, Mine T, et al. Recent advances in cancer vaccines: an overview. Japan J Clin Oncol. 2008;39(2):73–80.
- Klebanoff CA, Acquavella N, Yu Z, et al. Therapeutic cancer vaccines: are we there yet? Immunol Rev. 2011;239(1):27–44.
- Shumway NM, Ibrahim N, Ponniah S, et al. Therapeutic breast cancer vaccines: a new strategy for early-stage disease. BioDrugs. 2009;23(5):277–287.
- Liu SY, Wei W, Yue H, et al. Nanoparticles-based multi-adjuvant whole cell tumor vaccine for cancer immunotherapy. Biomaterials. 2013;34(33):8291–8300.
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10.
- Zamarin D, Ricca JM, Sadekova S, et al. PD-L1 in tumor microenvironment mediates resistance to oncolytic immunotherapy. J Clin Investig. 2018;128(11):5184.
- Ostman A. The tumor microenvironment controls drug sensitivity. Nat Med. 2012;18(9):1332–1334.
- Nahas MR, Rosenblatt J, Lazarus HM, et al. Anti-cancer vaccine therapy for hematologic malignancies: an evolving era. Blood Rev. 2018;32(4):312–325.
- Swartz MA, Iida N, Roberts EW, et al. Tumor microenvironment complexity: emerging roles in cancer therapy. Cancer Res. 2012;72(10):2473–2480.
- Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–322.
- Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21(4):687–692.
- Zi F, He J, He D, et al. Fibroblast activation protein alpha in tumor microenvironment: recent progression and implications (review). Mol Med Rep. 2015;11(5):3203–3211.
- Pleshkan VV, Alekseenko IV, Tyulkina DV, et al. Fibroblast activation protein (FAP) as a possible target of an antitumor strategy. Mol Genet Microbiol Virol. 2016;31(3):125–134.
- Liao D, Luo Y, Markowitz D, et al. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS One. 2009;4(11):e7965.
- Loeffler M, Kruger JA, Niethammer AG, et al. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Investig. 2006;116(7):1955–1962.
- Wen Y, Wang CT, Ma TT, et al. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer Sci. 2010;101(11):2325–2332.
- Chen M, Xiang R, Wen Y, et al. A whole-cell tumor vaccine modified to express fibroblast activation protein induces antitumor immunity against both tumor cells and cancer-associated fibroblasts. Sci Rep. 2015;5(1):14421.
- Fredriksen AB, Bogen B. Chemokine-idiotype fusion DNA vaccines are potentiated by bivalency and xenogeneic sequences. Blood. 2007;110(6):1797–1805.
- Huebener N, Fest S, Hilt K, et al. Xenogeneic immunization with human tyrosine hydroxylase DNA vaccines suppresses growth of established neuroblastoma. Mol Cancer Ther. 2009;8(8):2392–2401.
- Wei YQ, Wang QR, Zhao X, et al. Immunotherapy of tumors with xenogeneic endothelial cells as a vaccine. Nat Med. 2000;6(10):1160–1166.
- He QM, Wei YQ, Tian L, et al. Inhibition of tumor growth with a vaccine based on xenogeneic homologous fibroblast growth factor receptor-1 in mice. J Biol Chem. 2003;278(24):21831–21836.
- Strioga MM, Darinskas A, Pasukoniene V, et al. Xenogeneic therapeutic cancer vaccines as breakers of immune tolerance for clinical application: to use or not to use? Vaccine. 2014;32(32):4015–4024.
- Barnas JL, Simpson-Abelson MR, Yokota SJ, et al. T cells and stromal fibroblasts in human tumor microenvironments represent potential therapeutic targets. Cancer Microenviron. 2010;3(1):29–47.
- Cavallo F, Forni G. Recent advances in cancer immunotherapy with an emphasis on vaccines. Expert Rev Vac. 2009;8(1):25–28.
- Bi Y, Wei L, Mao HT, et al. Expressions of Fas, CTLA-4 and RhoBTB2 genes in breast carcinoma and their relationship with clinicopathological factors. Zhonghua Zhong Liu Za Zhi [Chin J Oncol]. 2008;30(10):749–753.
- Quandt D, Seliger B. “Tumor immunology meets oncology” (TIMO) X, May 23–24, 2014, Halle/Saale, Germany. Cancer Immunol Immunother. 2015;64(4):519–526.
- Vertuani S, Triulzi C, Roos AK, et al. HER-2/neu mediated down-regulation of MHC class I antigen processing prevents CTL-mediated tumor recognition upon DNA vaccination in HLA-A2 transgenic mice. Cancer Immunol Immunother. 2009;58(5):653–664.
- Grzelak A, Polakova I, Smahelova J, et al. Experimental combined immunotherapy of tumours with major histocompatibility complex class I downregulation. Int J Mol Sci. 2018;19(11):3693.
- Janakiram M, Abadi YM, Sparano JA, et al. T cell coinhibition and immunotherapy in human breast cancer. Discov Med. 2012;14(77):229–236.
- Vasievich EA, Huang L. The suppressive tumor microenvironment: a challenge in cancer immunotherapy. Mol Pharm. 2011;8(3):635–641.
- Gajewski TF, Woo SR, Zha Y, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. 2013;25(2):268–276.
- Guo G, Cui Y. New perspective on targeting the tumor suppressor p53 pathway in the tumor microenvironment to enhance the efficacy of immunotherapy. J Immunother Cancer. 2015;3(1):9.
- Buque A, Bloy N, Aranda F, et al. Trial watch-small molecules targeting the immunological tumor microenvironment for cancer therapy. Oncoimmunology. 2016;5:e1149674.
- Muntimadugu E, Kommineni N, Khan W. Exploring the potential of nanotherapeutics in targeting tumor microenvironment for cancer therapy. Pharmacol Res. 2017;126:109–122.
- Comes A, Rosso O, Orengo AM, et al. CD25+ regulatory T cell depletion augments immunotherapy of micrometastases by an IL-21-secreting cellular vaccine. J Immunol. 2006;176(3):1750–1758.
- Disis ML. Enhancing cancer vaccine efficacy via modulation of the tumor microenvironment. Clin Cancer Res. 2009;15(21):6476–6478.
- Sun Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016;380(1):205–215.
- Roma-Rodrigues C, Mendes R, Baptista PV, et al. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci. 2019;20:pii: E840.
- Alexander AN, Huelsmeyer MK, Mitzey A, et al. Development of an allogeneic whole-cell tumor vaccine expressing xenogeneic gp100 and its implementation in a phase II clinical trial in canine patients with malignant melanoma. Cancer Immunol Immunother. 2006;55(4):433–442.
- Xia Q, Zhang FF, Geng F, et al. Anti-tumor effects of DNA vaccine targeting human fibroblast activation protein alpha by producing specific immune responses and altering tumor microenvironment in the 4T1 murine breast cancer model. Cancer Immunol Immunother. 2016;65(5):613–624.
- Yi T, Wei YQ, Tian L, et al. Humoral and cellular immunity induced by tumor cell vaccine based on the chicken xenogeneic homologous matrix metalloproteinase-2. Cancer Gene Ther. 2007;14(2):158–164.
- Genoud N, Ott D, Braun N, et al. Antiprion prophylaxis by gene transfer of a soluble prion antagonist. Am J Pathol. 2008;172(5):1287–1296.
- Zhang G, Li J, Li S, et al. Exploring spatial trends and influencing factors for gastric cancer based on Bayesian statistics: a case study of Shanxi, China. Int J Environ Res Public Health. 2018;15.
- Turcotte S, Gros A, Hogan K, et al. Phenotype and function of T cells infiltrating visceral metastases from gastrointestinal cancers and melanoma: implications for adoptive cell transfer therapy. J Immunol. 2013;191(5):2217–2225.