
Abstract
Bronchial epithelial mitochondrial dysfunction including impaired mitochondrial biogenesis has been linked with the initiation and development of bronchial asthma. Montelukast, a robust antagonist of cysteinyl leukotriene receptors, has been widely applied for the therapies of bronchial asthma. However, the effects of montelukast in airway epithelial mitochondrial dysfunction are less reported. In the present study, we report that montelukast treatment in human bronchial epithelial cells of Beas-2b increased the expressions of PGC-1α, NRF-1 and TFAM. As expected, montelukast promoted mitochondrial biogenesis in Beas-2b cells through increasing mitochondrial mass, mtDNA/nDNA and the expression of cytochrome B. Importantly, we found that montelukast caused a functional gain in mitochondria of Beas-2b cells. Mechanistically, we found that montelukast treatment increased intracellular cAMP levels and activation of CREB. Blockage of CREB with H89 abolished montelukast-induced expression of PGC-1α. These findings report a novel pharmacological function of montelukast in stimulating mitochondrial biogenesis in Beas-2b cells, mediating by the CREB/PGC-1α pathway.
Introduction
Mitochondria are highly dynamic organelles that act as “energy factories” by generating intracellular adenosine triphosphate (ATP). Mitochondrial malfunction is important for bioenergetic metabolism and non-energetic pathological progression of various lung diseases, including bronchial asthma [Citation1]. Mitochondrial biogenesis has been reported to be important for sustaining normal mitochondrial homeostasis in various tissue and cell types. Human bronchial epithelial mitochondria provide an essential contribution to lung diseases. Notably, increasing evidence has shown that dysregulation of mitochondrial biogenesis is linked with a diversity of lung diseases, including bronchial asthma. PGC-1α acts as a central controller of mitochondrial biogenesis [Citation2]. In epithelial cells, activation of PGC-1α triggers the expression of NRF1 and TFAM. NRF1 drives nuclear genes to express mitochondrial proteins. TFAM is responsible for the expression of mitochondrial DNA (mtDNA) [Citation3]. Reduced expression of PGC-1α has been linked with the pathogenesis of bronchial asthma [Citation4]. Intervention of mitochondrial biogenesis via targeting PGC-1α has become an attractive approach for the therapy of epithelial dysfunction-associated lung diseases.
Montelukast, a selective antagonist of cysteinyl leukotriene (cysLT) receptor 1, is licenced for the therapy of bronchial asthma through blocking the action of leukotrienes [Citation5]. A diversity of pharmacological capacities of montelukast has been reported. Montelukast has displayed antioxidant and anti-inflammatory effects in various diseases. Montelukast has been shown to improve motor recovery and reduce IL-6 levels in spinal cord ischemia-reperfusion (I/R) injury [Citation6]. Another study demonstrated that administration of montelukast decreased respiratory syncytial virus-induced mucus hyperproduction in a rodent in vivo model [Citation7]. Interestingly, montelukast treatment has been reported to increase concentrations of intracellular cyclic adenosine monophosphate (cAMP) through inhibiting the activity of cyclic nucleotide phosphodiesterases [Citation5]. Consistently, another study reported that montelukast suppressed neutrophil-induced Ca2+-mediated pro-inflammatory activities of the cells in a cAMP dependent mechanism [Citation8]. cAMP plays a critical role in mediating activation of CREB [Citation9]. CREB has been shown to promote the expression of PGC-1α [Citation10]. These results suggest that montelukast might possess an impact on PGC-1α signalling and mitochondrial biogenesis.
Materials and methods
Cell culture and treatment
Human bronchial epithelial cells of Beas-2b were obtained from the Shanghai Cell Bank of Chinese Academy of Sciences. Cells were cultured in RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (Gibco, Carlsbad, CA, USA) and 1% antibiotics (penicillin/streptomycin; Sigma-Aldrich, St. Louis, MO, USA) in T75 cell culture flasks. Montelukast was from MedChemExpress, Princeton, NJ, USA and dissolved in dimethyl sulfoxide. To determine the effects of montelukast on PGC-1α expression, cells were stimulated with montelukast (5, 10 and 20 µM) [Citation11,Citation12] for 24 h in 6-well plates. To determine the effects of montelukast in NRF1 and TFAM, cells were stimulated with montelukast (10 µM) for 24 h.
Real-time PCR
RNA was extracted from Beas-2b cells using Trizol (Life Technologies, Carlsbad, CA, USA). The NanoDrop microvolume spectrophotometers were used to examine RNA concentration and quality. cDNA was generated using a RT-PCR with an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). Two microlitre of product was applied for real-time PCR analysis using SYBR Green master mix. The 2–ΔΔCt method was used to calculate target gene expressions by normalizing to GAPDH [Citation13].
Western blot analysis
Cells were homogenized and lysed with cell lysis buffer containing protease and phosphatase cocktail inhibitors (Sigma-Aldrich). A total of 20 µg lysates were separated on 10% SDS-PAGE and transferred to PVDF membranes (Bio-Rad Laboratories) [Citation14]. After blocked with 5% non-fat milk in in tris-buffered saline tween-20, membranes were incubated with primary antibodies overnight at 4 °C and horseradish peroxidase-conjugated secondary antibodies for additional 2 h. Blots were developed by enhanced chemiluminescence (Santa Cruz Biotechnology, Santa Cruz, USA). The bands of target protein were scanned and analysed with the image J software (NIH, Bethesda, MD, USA). Individual band in each experiment was selected and the background was subtracted. Integrated signal intensities for each band was quantified to reflect the amount of the protein level. Results were normalized to β-actin.
MitoTracker red staining
Mitochondrial mass in Beas-2b epithelial cells was examined by staining with MitoTracker red. After stimulation, Beas-2b cells were probed with 20 nM MitoTracker red (Thermo Fisher Scientific) for 30 min at 37 °C without light. Nuclei of Beas-2b cells were counterstained with DAPI (Thermo Fisher Scientific). Fluorescent images were visualized and recorded using a fluorescence microscope. The average integrated optical density (IOD) of 100 randomly selected cells from each group was calculated and used to represent mitochondrial mass. The software Image J was used to quantify the average IOD in epithelial cells of Beas-2b. First, regions of interest (ROI) in the fluorescent images were defined. Second, the number of Beas-2b epithelial cells was counted. Third, the integrated density value (IDV) in ROI was determined. The average IOD = IDV/Cell number.
Measurement of mtDNA
The mtDNA in Beas-2b epithelial cells was examined by real-time PCR analysis as described above. Total DNA was isolated from Beas-2b epithelial cells using a QIAamp DNA mini kit (QIAGEN, CA, USA). NanoDrop microvolume spectrophotometers were used to measure the concentration of DNA. The mitochondria-derived gene ND1 and the nucleus-derived gene 18S were used to index mitochondrial and nuclear DNA, respectively. mtDNA/nDNA was calculated.
Detection of mitochondrial respiratory rate
To clarify the effect of montelukast treatment in mitochondrial function, the O2 consumption of Beas-2b cells was assessed using a respirometer (Oroboros Instrument, Danvers, MA, USA). Briefly, 5 × 106 Beas-2b cells were collected and resuspended in 2 mL RPMI 1640 medium and then added into a glass chamber equilibrated in ambient room air. Samples were stirred at 750 rpm for 10 min. The oxygen content in the reaction system was recorded at 2 s intervals.
Determination of intracellular ATP production
ATP levels in Beas-2b cells were assessed using a commercial firefly luciferase ATP detection kit (Beyotime, China). Briefly, Beas-2b cells were collected, lysed and centrifuged at 10,000 × g. Fifty microlitre of supernatant was mixed with equal volume of the luciferin/luciferase reagent. Chemiluminescence was immediately detected using a fluorescence microplate reader.
Measurement of intracellular cAMP
A total of 1 × 107 Beas-2b cells were stimulated with 10 µM montelukast for 1 h. Cells were washed with ice-cold HBSS and harvested. After a brief centrifugation at 500 × g, 1 mL of 0.1 M HCl was used to extract cAMP from pellets of Beas-2b cells. The intracellular cAMP concentration was assayed as previously described [Citation8].
Statistical analysis
Experimental data are presented as means ± SEM Statistical analysis was performed using ANOVA using SPSS (Version 20.0), followed by the Bonferroni’s post-hoc test. p < .05 was considered statistically significant.
Results
Montelukast treatment activates the PGC-1α-NRF1-TFAM signalling pathway
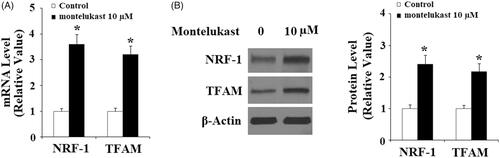
Here, we found that montelukast treatment at 5, 10 and 20 µM led to a sustainable elevation of PGC-1α () in Beas-2b cells. Notably, we found that 10 µM montelukast obviously increased NRF1 and TFAM at the gene levels (). Accordingly, 10 µM montelukast treatment also upregulated the expression of NRF1 and TFAM at the protein levels ().
Figure 1. Montelukast treatment increased the expression of PGC-1α in Beas-2b cells. (A–B) Beas-2b cells were stimulated with montelukast (5, 10, and 20 µM) for 24 h. Expression of PGC-1α was measured (*, p < .01 vs. vehicle group; #, p < .01 vs. 5 µM montelukast group; $, p < .01 vs. 10 µM montelukast group, n = 5–6).
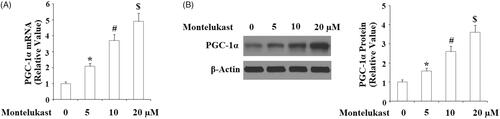
Figure 2. Montelukast treatment increased the expression of NRF-1 and TFAM in Beas-2b cells. (A–B) Beas-2b cells were treated with 10 µM montelukast for 24 h. Expression of NRF-1 and TFAM was measured (*, p < .01 vs. vehicle group, n = 5–6).
Montelukast treatment promotes mitochondrial biogenesis
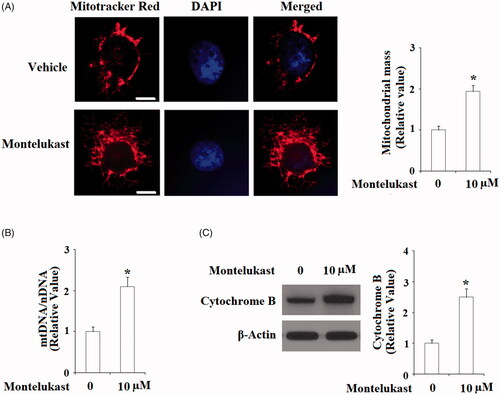
To assess the effects of montelukast in mitochondrial biogenesis, mitochondrial mass of Beas-2b cells was examined using MitoTracker red staining. As shown in , 10 µM montelukast treatment significantly increased MitoTracker red staining, suggesting that montelukast has an important impact in increasing mitochondrial mass. Induction of mtDNA expression mediated by TFAM is an essential event of mitochondrial biogenesis. Here, our findings indicate that the presence of montelukast significantly increased mtDNA/nDNA (). The mitochondrial protein cytochrome B is an important respiratory subunit. Consistently, montelukast remarkably upregulated the expression of cytochrome B ().
Figure 3. Montelukast treatment promoted mitochondrial biogenesis in Beas-2b cells. Cells were stimulated with 10 µM montelukast for 24 h. (A) Mitochondrial mass; (B) mtDNA/nDNA; (C) Expression of cytochrome B (*, p < .01 vs. vehicle group, n = 5–6).
Montelukast treatment causes “gain of mitochondrial function” in Beas-2b cells
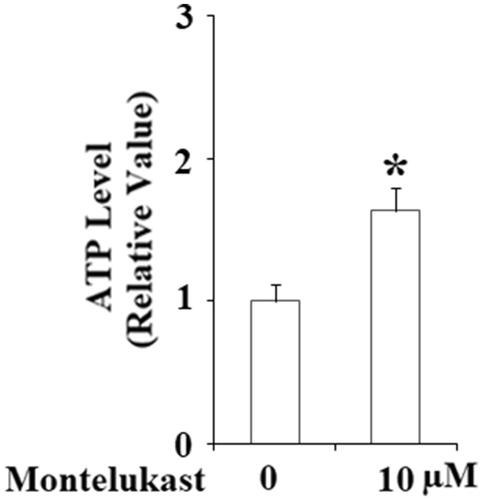
The stimulatory effects of montelukast in mitochondrial biogenesis implicate that montelukast might have a positive impact on mitochondrial function in Beas-2b cells. Hence, we evaluated the effect of montelukast exposure on mitochondrial function by measuring mitochondrial respiration and ATP production. Results demonstrated that oxygen consumption in montelukast-treated cells was remarkably higher than that in non-treatment cells (). As shown in , montelukast treatment increased the respiratory rate of Beas-2b cells from 16.3 ± 1.8 to 24.9 ± 2.5 [pmol/(s × Mill)]. Importantly, montelukast treatment significantly increased ATP generation in Beas-2b cells (). These results implicate that montelukast induced a gain of mitochondrial function in human bronchial epithelial cells.
Figure 4. Montelukast treatment induced a gain in mitochondrial function in Beas-2b cells. Cells were stimulated with 10 µM montelukast for 24 h. (A). Representative results of oxygen content consumption in non-treated (blue curve) and montelukast-treated (purple curve) Beas-2b cells; (B). Summarized mitochondrial respiratory rate (*, p < .01 vs. vehicle group, n = 6).
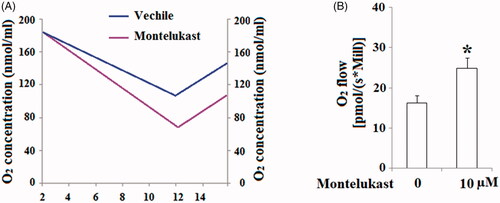
Figure 5. Montelukast treatment increased the production of ATP in Beas-2b cells. Cells were stimulated with 10 µM montelukast for 24 h. ATP production was measured (*, p < .01 vs. vehicle group, n = 6).
The stimulatory effects of montelukast on PGC-1α expression require the activation of CREB
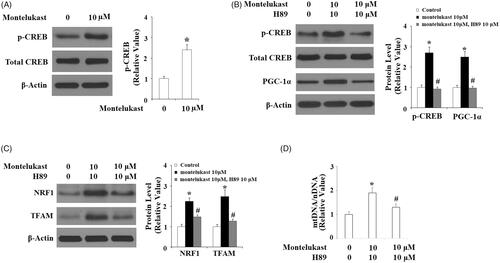
Intracellular cAMP acts as a second messenger and promotes activation of the transcriptional factor CREB. Activation of CREB is an essential step to initiate the transcription of PGC1-α [Citation15]. Here, montelukast treatment obviously increased intracellular levels of cAMP (). Accordingly, montelukast enhanced the phosphorylation of CREB at Ser133 but did not change total CREB (). To confirm the participation of CREB in mediating montelukast-induced expression of PGC-1α, cells were stimulated with 10 µM montelukast with or without H89 for 24 h. Results in indicate that H89 completely abolished montelukast-induced expression of PGC-1α, suggesting the involvement of CREB. Notably, H89 inhibited montelukast-mediated expression of NRF1 and TFAM (), two executor proteins of mitochondrial biogenesis. Correspondingly, H89 reduced montelukast-mediated expression of mtDNA/nDNA (). These findings reveal that the effects of montelukast in promoting mitochondrial biogenesis are mediated by CREB.
Figure 6. Montelukast-induced generation of cAMP in Beas-2b cells. Cells were stimulated with 10 µM montelukast for 1 h. Intracellular levels of cAMP were determined (*, p < .01 vs. vehicle group, n = 6).
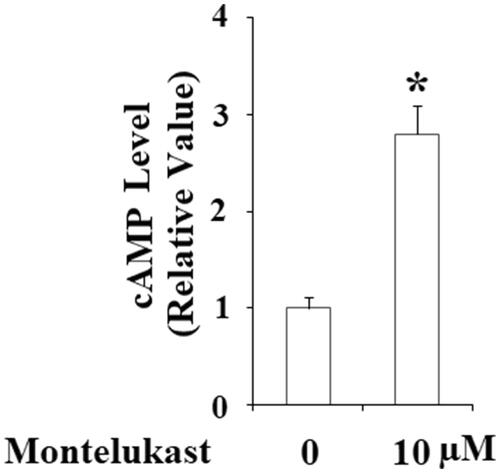
Figure 7. Montelukast-induced expression of PGC-1α in Beas-2b cells is mediated by cAMP/CREB. (A) Beas-2b cells were stimulated with 10 µM montelukast for 1 h. Phosphorylated and total levels of CREB were determined; (B) Cells were stimulated with 10 µM montelukast with or without H89 (10 μM) for 24 h. The expressions of phosphorylated CREB, total CREB and PGC-1α were measured by western blot analysis; (C) Beas-2b cells were stimulated with 10 µM montelukast with or without H89 (10 μM) for 24 h. The expressions of NRF1 and TFAM were measured by western blot analysis; (D) Beas-2b cells were stimulated with 10 µM montelukast with or without H89 (10 μM) for 24 h. mtDNA/nDNA was determined (*, #, p < .01 vs. previous column group, n = 6).
Discussion
CysLTs are linked with the pathogenesis of asthma. As an important anti-leukotriene compound, montelukast has displayed its efficacy and safety profile in different age teams with asthma [Citation16]. Montelukast has a significant inhibitory effect against bronchoconstriction caused by leukotriene D4 [Citation17]. Administration of montelukast plays important roles in improving pulmonary function and reducing the risk of exacerbation in patients with asthma. In addition to its bronchoprotective effect, montelukast has exhibited anti-inflammatory and anti-allergic properties in various tissues and cells [Citation18]. However, the physiological function of montelukast on mitochondrial function in human bronchial epithelial cells remains undefined. Here, we identified a novel pharmacological function of montelukast in promoting mitochondrial biogenesis in Beas-2b epithelial cells. First, montelukast treatment stimulated the expression of the “master switch” molecules of mitochondrial biogenesis, PGC-1α, NRF1 and TFAM. Second, montelukast treatment increased mitochondrial mass, mtDNA/nDNA and the expression of cytochrome B. Third, the presence of montelukast led to a functional gain for mitochondria by enhancing the mitochondrial respiratory rate and ATP generation. Mechanistically, we revealed that activation of CREB through elevation of intracellular cAMP participated in montelukast-induced expression of PGC-1α and blockage of CREB activation, while H89 abolished this effect.
Mitochondrial deterioration has been linked with the pathological development of various diseases, including chronic pulmonary diseases [Citation19]. In addition, mitochondrial malfunction plays an important role in bronchial epithelial dysfunction, which is involved in the initiation and progression of several lung diseases, such as asthma, asbestosis and tumorigenesis [Citation4]. The induction of mitochondrial biogenesis in epithelial cells is essential for activating mitochondrial quality control and promoting cell survival in support of alveolar function in rodent pneumonia models [Citation20]. Reduced PGC-1α expression and dysregulation of mitochondrial biogenesis in alveolar epithelial cells have been shown to mediate TGF-β-induced pulmonary fibrosis [Citation21]. Therefore, preserving PGC-1α expression and mitochondrial function in bronchial epithelial cells via administration of montelukast may serve as a novel mechanism for slowing down the pathogenesis of chronic lung diseases. Transcriptional modulation of PGC-1α is complex. The transcriptional factor CREB is shown to be a key regulator of PGC-1α transcription [Citation22]. In the present study, we display that the effect of montelukast on PGC-1α expression is mediated by cAMP/CREB signalling. Consistently, montelukast treatment can induce intracellular cAMP synthesis [Citation23]. In addition to regulating mitochondrial biogenesis, diverse and new physiological functions of PGC-1α in regulating lipid metabolism, energy expenditure, the inflammatory response, and oxidative stress have been reported in recent studies [Citation2]. These findings suggest that montelukast might possess a wide range of pharmacological capacities via modulation of PGC-1α.
To sum up, we report a novel pharmacological function of montelukast in regulating mitochondrial biogenesis in human bronchial epithelial cells through promoting the activation of PGC-1α signalling.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Schumacker PT, Gillespie MN, Nakahira K, et al. Mitochondria in lung biology and pathology: more than just a powerhouse. Am J Physiol Lung Cell Mol Physiol. 2014;306(11):L962–L974.
- Villena JA. New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015;282(4):647–672.
- Zhang Y, Xu H. Translational regulation of mitochondrial biogenesis. Biochem Soc Trans. 2016;44(6):1717–1724.
- Zhang L, Wang W, Zhu B, et al. Epithelial mitochondrial dysfunction in lung disease. Adv Exp Med Biol. 2017;1038:201–217.
- Theron AJ, Steel HC, Tintinger GR, et al. Cysteinyl leukotriene receptor-1 antagonists as modulators of innate immune cell function. J Immunol Res. 2014;2014:608930.
- Korkmaz K, Gedik HS, Budak AB, et al. Effect of montelukast on spinal cord ischemia- reperfusion injury. Turk Neurosurg. 2015;25(5):757–765.
- Han J, Jia Y, Takeda K, et al. Montelukast during primary infection prevents airway hyperresponsiveness and Inflammation after reinfection with respiratory syncytial virus. Am J Respir Crit Care Med. 2010;182(4):455–463.
- Anderson R, Theron AJ, Gravett CM, et al. Montelukast inhibits neutrophil pro-inflammatory activity by a cyclic AMP-dependent mechanism. Br J Pharmacol. 2009;156(1):105–115.
- Casaburi I, Avena P, Lanzino M, et al. Chenodeoxycholic acid through a TGR5-dependent CREB signaling activation enhances cyclin D1 expression and promotes human endometrial cancer cell proliferation. Cell Cycle. 2012;11(14):2699–2710.
- Lonard DM, O'Malley BW. The expanding cosmos of nuclear receptor coactivators. Cell. 2006;125(3):411–414.
- Dong H, Liu F, Ma F, et al. Montelukast inhibits inflammatory response in rheumatoid arthritis fibroblast-like synoviocytes. Int Immunopharmacol. 2018;61:215–221.
- Di X, Tang X, Di X. Montelukast inhibits oxidized low-density lipoproteins (ox-LDL) induced vascular endothelial attachment: an implication for the treatment of atherosclerosis. Biochem Biophys Res Commun. 2017;486(1):58–62.
- Rosenberg N, Rosenberg O, Weizman A, et al. In vitro effect of FGIN-1-27, a ligand to 18 kDa mitochondrial translocator protein, in human osteoblast-like cells. J Bioenerg Biomembr. 2014;46(3):197–204.
- Zhang P, Ma J, Gao J, et al. Downregulation of monocarboxylate transporter 1 inhibits the invasion and migration through suppression of the PI3K/Akt signaling pathway in human nasopharyngeal carcinoma cells. J Bioenerg Biomembr. 2018;50(4):271–281.
- Herzig S, Long F, Jhala US, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413(6852):179–183.
- Marcello C, Carlo L. Asthma phenotypes: the intriguing selective intervention with montelukast. Asthma Res Pract. 2016;2:11.
- Kar M, Altıntoprak N, Muluk NB, et al. Antileukotrienes in adenotonsillar hypertrophy: a review of the literature. Eur Arch Otorhinolaryngol. 2016;273(12):4111–4117.
- Barbosa JS, Almeida Paz FA, Braga SS. Montelukast medicines of today and tomorrow: from molecular pharmaceutics to technological formulations. Drug Deliv. 2016;23(9):3257–3265.
- Nam HS, Izumchenko E, Dasgupta S, et al. Mitochondria in chronic obstructive pulmonary disease and lung cancer: where are we now? Biomarkers Med. 2017;11(6):475–489.
- Suliman HB, Kraft B, Bartz R, et al. Mitochondrial quality control in alveolar epithelial cells damaged by S. aureus pneumonia in mice. Am J Physiol Lung Cell Mol Physiol. 2017;313(4):L699–L709.
- Sohn EJ, Kim J, Hwang Y, et al. TGF-β suppresses the expression of genes related to mitochondrial function in lung A549 cells. Cell Mol Biol (Noisy-le-grand). 2012;Suppl.58:OL1763-7.
- Louet JF, Hayhurst G, Gonzalez FJ, et al. The coactivator PGC-1 is involved in the regulation of the liver carnitine palmitoyltransferase I gene expression by cAMP in combination with HNF4 alpha and cAMP-response element-binding protein (CREB). J Biol Chem. 2002;277(41):37991–38000.
- Fogli S, Stefanelli F, Neri T, et al. Montelukast prevents microparticle-induced inflammatory and functional alterations in human bronchial smooth muscle cells. Pharmacol Res. 2013;76:149–156.